


各位医生大大们,你们是否在医学研究的海洋中奋力前行,寻找着那些能够揭示疾病奥秘的钥匙呢?今天,我要给你们介绍一对超强组合——孟德尔随机化和生信分析,它们就像是医学研究中的“神雕侠侣”,携手为我们带来更多的发现和突破。
首先,咱们来聊聊生信分析。这可是个厉害的家伙,它能把那些复杂的生物数据变得有条有理,就像是把一团乱麻梳理成整齐的丝线。生信分析通过对基因组、转录组、蛋白质组等数据的分析,可以帮助我们找到疾病的生物标志物,揭示疾病的发生机制,为精准医疗提供有力的支持。
而孟德尔随机化呢,则像是一个公正的裁判,它利用遗传学数据来推断因果关系,避免了传统观察性研究中混杂因素和反向因果关系的干扰。就像是在一场比赛中,孟德尔随机化能够确保评判的公正性,让我们更准确地了解暴露因素与疾病结局之间的真正关系。
那么,孟德尔随机化和生信分析结合起来会产生怎样的火花呢?这就像是给生信分析加上了一双翅膀,让它能够飞得更高更远。它们的结合可以更深入地挖掘生物数据中的信息,为医学研究提供更可靠的证据。
比如说,在研究某种疾病的危险因素时,生信分析可以找出一些潜在的关联,而孟德尔随机化则可以进一步验证这些关联是否真的存在因果关系。这样一来,我们就能更准确地判断哪些因素是导致疾病的真正元凶,从而制定更有效的治疗和预防策略。
而且,这种结合还能帮助我们发现一些隐藏在数据背后的秘密。就像是在玩一场寻宝游戏,孟德尔随机化和生信分析一起帮我们找到那些珍贵的宝藏,为医学研究带来更多的惊喜。
所以,医生们,不妨试试将孟德尔随机化和生信分析结合起来,让它们为你们的研究助力。相信它们会给你们带来意想不到的收获,让你们在医学研究的道路上更加得心应手。
快来加入这场奇妙的医学研究之旅吧,让孟德尔随机化和生信分析成为你们的得力助手,一起揭开疾病的神秘面纱!
《探索医学新视角:医学生信分析的力量与发展趋势》
在医学领域不断发展的今天,医学生信分析正逐渐成为一颗璀璨的新星。
生信分析,即生物信息学分析,是一门结合了生物学、计算机科学和统计学的交叉学科。它主要通过对生物数据的收集、整理、分析和解释,来揭示生命现象的本质和规律。
在医学研究中,生信分析可以帮助我们更好地理解疾病的发生机制、诊断和治疗方法。例如,通过对基因组数据的分析,我们可以发现与疾病相关的基因变异,为精准医疗提供依据;通过对蛋白质组学数据的研究,我们可以了解蛋白质的功能和相互作用,寻找新的药物靶点。通过对海量的生物医学数据进行挖掘和分析,生信分析可以揭示疾病发生发展的潜在机制。从基因层面到蛋白质组学,从细胞信号通路到临床表型,它如同一位智慧的侦探,抽丝剥茧般地找出疾病的关键线索。
对于临床医生而言,医学生信分析提供了更精准的诊断工具。它可以帮助医生快速准确地判断疾病类型,制定个性化的治疗方案。例如,通过分析患者的基因信息,医生可以确定最适合的药物和治疗剂量,提高治疗效果,减少副作用。
在科研领域,医学生信分析更是发挥着举足轻重的作用。它为医学研究提供了新的思路和方法,加速了科研成果的转化。研究人员可以利用生信分析技术,对大规模的临床数据进行整合和分析,发现新的疾病标志物和治疗靶点,为新药研发提供有力支持。
**实际应用案例
以癌症研究为例,医学生信分析在肿瘤的诊断和治疗中发挥了巨大作用。通过对肿瘤患者的基因组数据进行分析,可以发现特定的基因突变与肿瘤的发生发展密切相关。例如,在乳腺癌中,BRCA1 和 BRCA2 基因突变与患者的患病风险增加有关。医生可以根据患者的基因检测结果,为其提供个性化的筛查和预防建议。此外,医学生信分析还可以帮助研究人员寻找新的肿瘤治疗靶点。通过对肿瘤细胞的转录组学和蛋白质组学数据进行分析,发现了一些在肿瘤细胞中高表达的蛋白质,这些蛋白质可能成为潜在的治疗靶点。目前,已经有一些针对这些靶点的药物正在研发中。
再比如,在传染病研究中,医学生信分析可以帮助追踪病毒的传播路径和变异情况。在新冠疫情期间,各国科学家利用生物信息学技术对新冠病毒的基因组进行测序和分析,了解病毒的变异情况和传播规律,为疫情防控提供了重要的科学依据。
**医学生信分析的发展趋势
• 多组学整合分析成为主流:单一的基因组学或蛋白质组学数据已经不能满足医学研究的需求,未来医学生信分析将越来越多地整合多组学数据,如基因组学、转录组学、蛋白质组学、代谢组学等,从多个层面全面揭示疾病的发生发展机制。例如,通过对肿瘤患者的多组学数据进行综合分析,可以更准确地找到肿瘤的驱动基因和关键分子,为精准治疗提供依据。
• 人工智能与机器学习的深度融合:人工智能和机器学习技术将在医学生信分析中得到更广泛的应用。它们可以帮助处理和分析海量的生物医学数据,挖掘潜在的模式和规律,提高分析的准确性和效率。例如,利用深度学习算法可以对医学影像数据进行分析,辅助医生进行疾病诊断;通过机器学习模型可以预测药物的疗效和不良反应等。
• 个性化医疗的推动:随着人们对健康的重视和医疗技术的不断进步,个性化医疗将成为未来医学的发展方向。医学生信分析可以为个性化医疗提供重要的支持,通过对患者的基因、蛋白质、代谢等多组学数据进行分析,为患者制定个性化的治疗方案,提高治疗效果和患者的生活质量。
• 与临床实践的紧密结合:未来,医学生信分析将更加紧密地与临床实践相结合,为临床决策提供支持。例如,通过对临床电子病历数据的分析,可以预测患者的疾病风险、治疗反应和预后,帮助医生制定更合理的治疗策略;利用生信分析技术对医疗大数据进行挖掘,可以发现疾病的流行趋势和危险因素,为公共卫生决策提供依据。
• 数据共享与合作的加强:生物医学数据的积累是医学生信分析的基础,未来数据共享和合作将变得更加重要。各国政府、科研机构和企业将加强合作,建立更大规模的生物医学数据库,并制定数据共享的标准和规范,促进数据的交流和利用。同时,跨学科的合作也将不断加强,生物信息学、医学、计算机科学等领域的专家将共同开展研究,推动医学生信分析的发展。
此外,医学生信分析还具有广阔的应用前景。随着技术的不断发展,生信分析将更加智能化、高效化。它可以实现疾病的早期预测和风险评估,为人们的健康管理提供科学依据。总之医学生信分析作为医学领域的前沿技术,正以其强大的力量推动着医学的进步。然而,生信分析需要专业的知识和技能,对于许多医生朋友来说,可能会面一些挑战。这时,选择一家专业的生信分析服务提供商就显得尤为重要。
森煜智研拥有一支专业的生信分析团队,具备丰富的经验和专业的知识,能够为您提供高质量的医学生信分析服务。无论是基因组学、转录组学、蛋白质组学还是代谢组学等方面的分析,森煜智研都能满足您的需求。
选择森煜智研代做医学生信分析,您将获得以下优势:
1. 专业的团队:确保分析结果的准确性和可靠性。
2. 个性化的服务:根据您的研究需求,定制专属的分析方案。
3. 高效的沟通:及时反馈分析进展,解答您的疑问。
4. 优质的售后:为您提供后续的支持和帮助。
如果您正在进行医学研究,不妨考虑让森煜智研为您的研究助力,共同揭开医学奥秘的钥匙。
本研究主要使用不同的生化和生物物理方法检验了玫瑰红嗜热球菌 N-去甲基化酶与嗜温枯草芽孢杆菌的 N-脱甲基酶的高热稳定性的性质,有助于理解嗜热酶的天然稳定框架,为工业应用中构建稳健酶提供参考。去甲基化是许多天然产物转化为活性形态的关键步骤,但是-C-N-键极为稳定,氮原子含有未成键的孤电子对,环上电子云密度大,碳氮键键长短,难以极化,且相关反应效率低,使得含氮基团较难去除,因此N-甲基脱除反应是目前有机合成的一大挑战。在团队前期研究工作中发现了一种来自玫瑰红嗜热球菌(Thermomicrobium roseum DSM 5159)的N-甲基脱除酶,即肌氨酸氧化酶(Sarcosine oxidase, EC 1.5.3.1, TrSOX),属于黄素蛋白氧化酶类,以FAD为辅因子,该酶针对N-甲基具有高效的生物脱除能力,并且具有一定的手性选择性,表现出优异的耐热性和环境抗性。
该研究指出几个影响酶热稳定性的关键因素,包括二硫键、盐桥、疏水性、氢键、螺旋含量和柔韧性,以及含有脯氨酸等刚性侧链的残基被认为在酶的稳定性中起着关键作用。此外,在蛋白质或基于蛋白质的药物产品的开发中,聚集或聚集相关的沉淀一直被认为是主要挑战之一,它会导致生物功能的丧失。
该研究基于玫瑰红嗜热球菌N-甲基脱除酶TrSOX的晶体结构,在聚集界面和刚性位点上引入取代,以降低聚集比和刚性。在聚集界面上,V162S、M308S、F170S和V306S的4个取代热稳定性显著降低,但催化效率有一定程度的提升。此外,耐热性框架在几个多重P→G的取代(P129G/P134G, P237G/P259G和P259G/P276G)中被严重破坏。这些结构波动与分子动力学模拟中的整体结构和局部的RMSD、氢键旋转半径和溶剂可溶表面积值具有良好的一致性。为了进一步确认TrSOX上聚集界面和脯氨酸残基的作用,将这些关键位点引入枯草芽孢来源的肌氨酸氧化酶,这些取代提高了Tm和ΔH值,氢键数增加,降低了RSMD、Rg和SASA值,促进热稳定性。尤其是突变体A249P、F309V、E161V和N238P,它们在60°C下的半衰期从小于10 min显著增加到1440 min、996min、640min和60min。
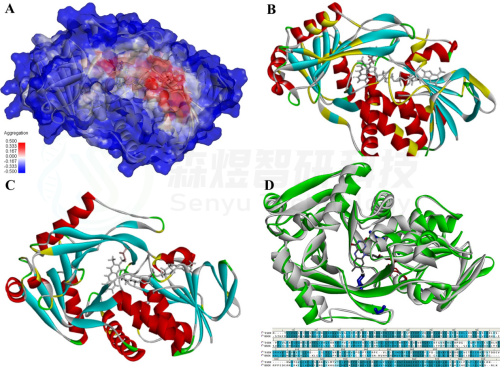
Figure 1. Structural design. (A) Aggregation interface of TrSOX. (B) The substitutions were designed to reduce the aggregation ratio on the aggregation interface (marked in yellow). (C) Flexible substitutions were designed at the surface proline sites (marked in yellow). (D) Structural and sequential alignments of TrSOX (green) and BSOX (white), and they shared ∼36.6% sequence identity.
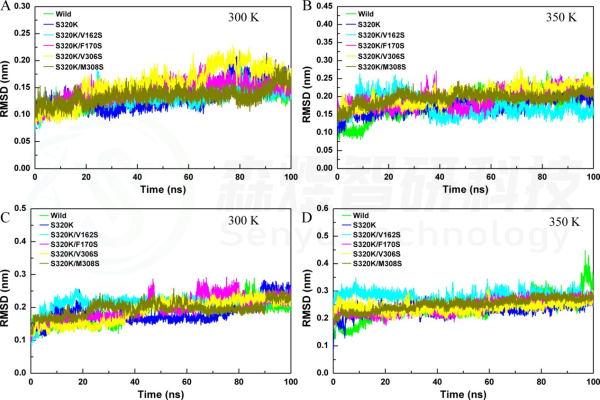
Figure 2. The RMSD values of substitutions on the aggregation interface. Whole-structure RMSD values of substitutions at (A) 300 K and (B) 350K. Substrate pocket RMSD of substitutions at(C)300K and(D)350K.
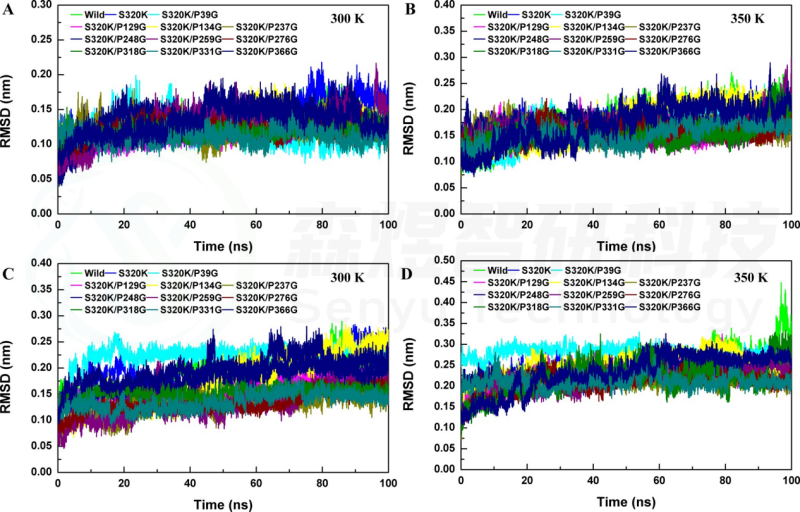
Figure 3. RMSD values of substitutions at proline sites. Whole-structure RMSD values of substitutions at (A) 300 K and (B) 350 K. Substrate pocket RMSD values of substitutions at (C) 300 K and (D) 350 K.

Figure 4. RMSD values of S320K/P129G/P134G. Whole structure RMSD values of substitutions at (A) 300 K and (B) 350 K. Partial RMSD (129−134) RMSD values of substitutions at (C) 300 K and (D) 350 K. Substrate pocket RMSD values of substitutions at (E) 300 K and (F) 350K. (G) Location of the fragment (129−134, marked in yellow).
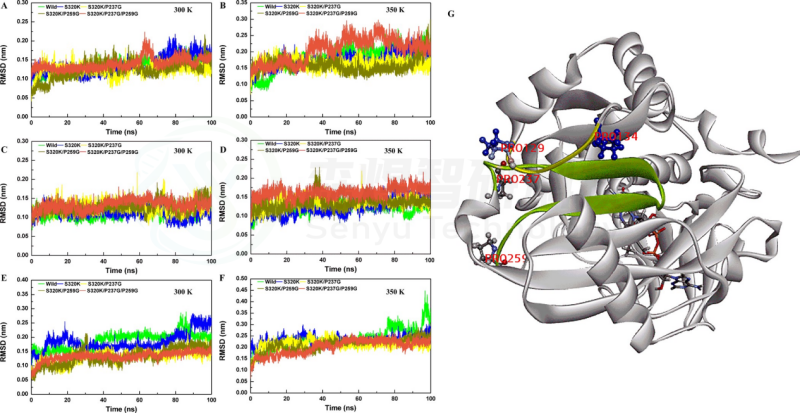
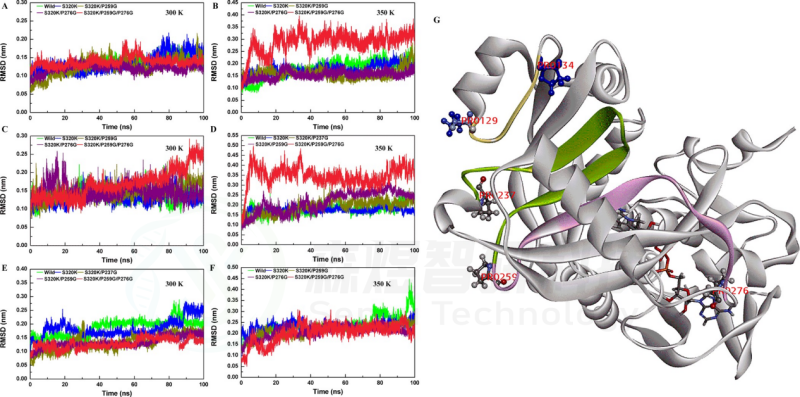
Figure 5. RMSD values of S320K/P237G/P259G. Whole-structure RMSD values of substitutions at (A) 300 K and (B) 350 K. Partial RMSD (237−259) values of substitutions at (C) 300 K and (D) 350 K. Substrate pocket RMSD values of substitutions at (E) 300 K and (F) 350 K. (G) Location of the fragment (129−134, marked in yellow; 237−259, marked in green).
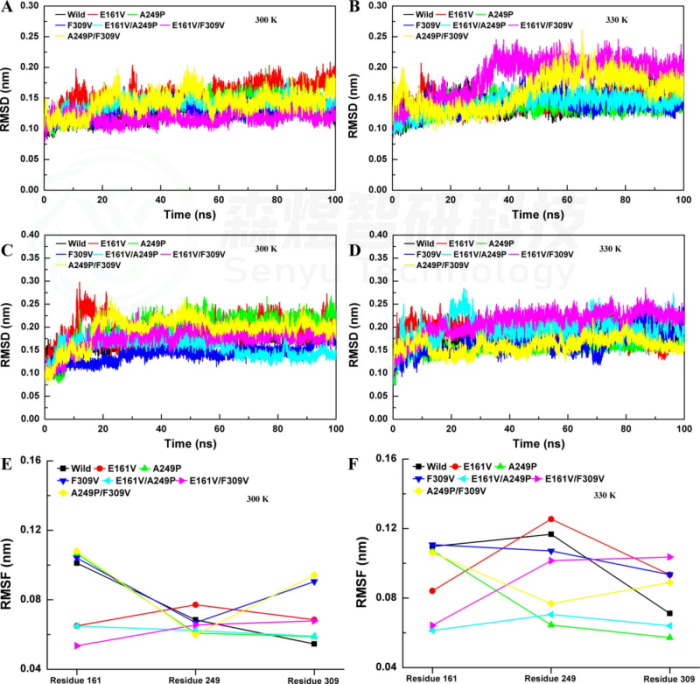
Figure 6. RMSD values of S320K/P259G/P276G. Whole-structure RMSD values of substitutions at (A) 300 K and (B) 350 K. Partial RMSD (259−276) values of substitutions at (C) 300 K and (D) 350 K. Substrate pocket RMSD values of substitutions at (E) 300 K and (F) 350 K. (G) (129−134, marked in yellow; 237−259, marked in green; 259−276, marked in pink).
原名:Aggregation Interface and Rigid Spots Sustain the Stable Framework of a Thermophilic
N-Demethylase
期刊:Journal of Agricultural and Food Chemistry
DOI号: 10.1021/acs.jafc.3c00877
18817128943

扫码关注 森煜智研微信号

扫码关注 森煜智研公众号