


以产气荚膜梭菌α毒素蛋白三维结构(PDB代码为1CA1[1])作为模板,根据客户提供的产气荚膜梭菌α毒素蛋白(CPA)结构的完整序列,将其与1CA1蛋白序列比对,结果如图1所示,两个蛋白间的序列一致性为99.46%。随后,采用SWISS-MODEL程序[2]对CPA蛋白进行同源建模,从而获取可用于后续分子识别研究的CPA蛋白三维结构模型,建模得到的蛋白结构如图2a所示。

图1CPA蛋白与1CA1蛋白序列比对结果
用PROCHECK[3]程序对补全并优化后的蛋白模型进行了评价。Ramachandran plot用于阐述蛋白质或肽立体结构中肽键内α碳原子和羰基碳原子间的键旋转度对α碳原子和氮原子间的键的旋转度,主要用来指明蛋白质或肽类中氨基酸残基的允许和不允许的构象。Ramachandran plot主要分为三个区域,完全允许区(红色区域),允许区(黄色区域),不允许区(空白区域)。图2b给出了CPA蛋白的Ramachandran plot结果,位于完全允许区的氨基酸占比为91.8%,位于允许区的氨基酸占比为7.3%,总和超过了99%,没有氨基酸残基位于扭转角禁止区域,因此表明建模所得CPA蛋白所有氨基酸残基的二面角均在合理范围内,符合立体化学能量规则。
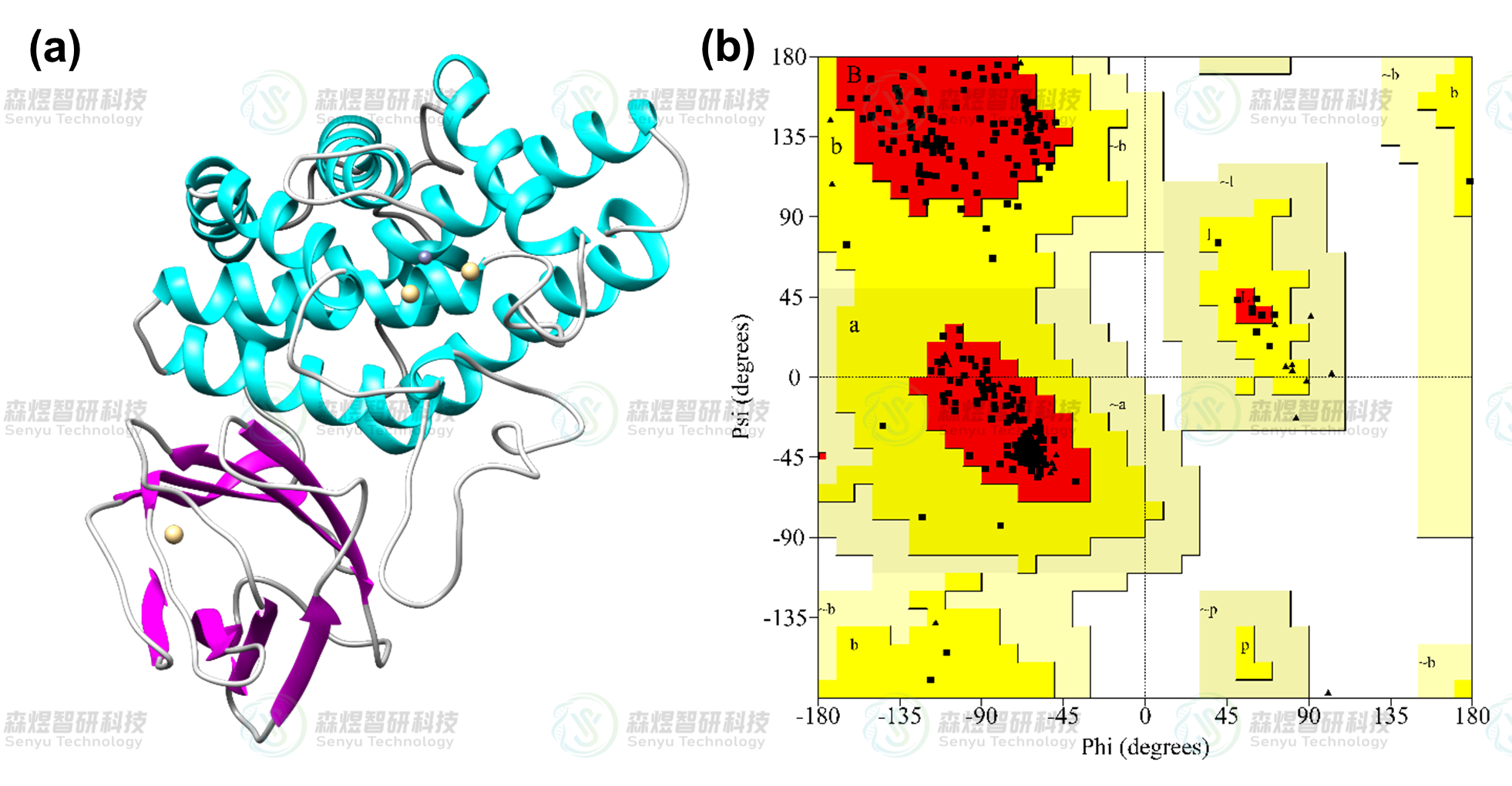
图2CPA蛋白结构(a)和CPA蛋白模型的Ramachandran Plot(b)
将同源建模得到的CPA蛋白作为初始结构用于后续的分子对接。初始结构用AutoDock Tools 1.5.6[4]进行处理,保存蛋白原有电荷,并生成pdbqt文件用于对接。
杨梅黄酮(MYR)底物分子结构从PubChem数据库下载,使用MOPAC程序[5]优化分子结构并计算PM3原子电荷[6]用于后续分子对接,采用AutoDock Tools 1.5.6处理配体结构,生成pdbqt文件用于对接。
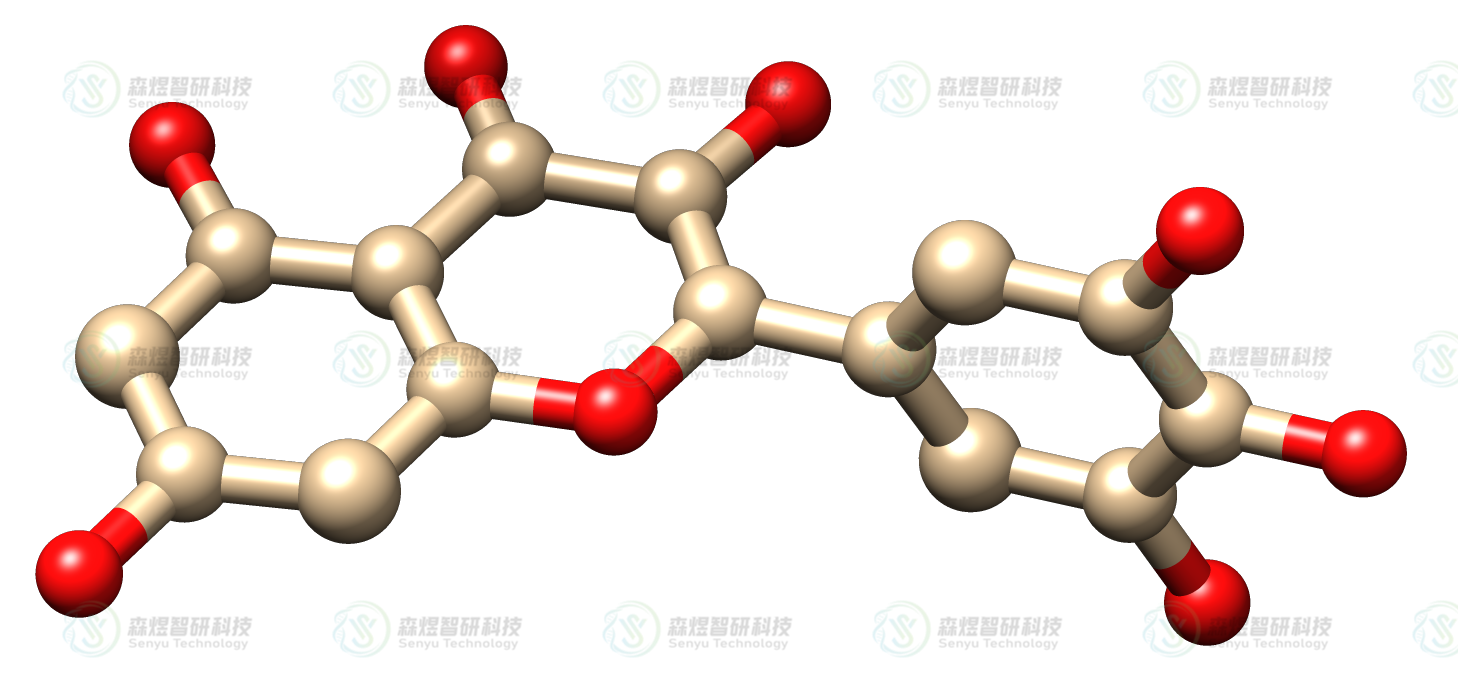
图3MYR分子的结构
通过文献调研发现,分子MYR在CPA蛋白上的结合位点位于蛋白中几段α-Helix结构围成空腔上方,空腔底部存在一个Zn2+离子和两个Cd2+离子(如图4所示),有利于分子与CPA蛋白中的活性空腔结合,占据底物的结合位点,抑制该蛋白的活性。因此本文将MYR分子与蛋白的结合位点也定于此口袋,使其能竞争性抑制蛋白的活性。分子对接采用AutoDock 4.2.6软件包实现,对接盒子中心坐标设为(12.701,19.827,28.957),XYZ各方向的格点数设为60×60×60,对接次数设为100,其余参数采用默认值。
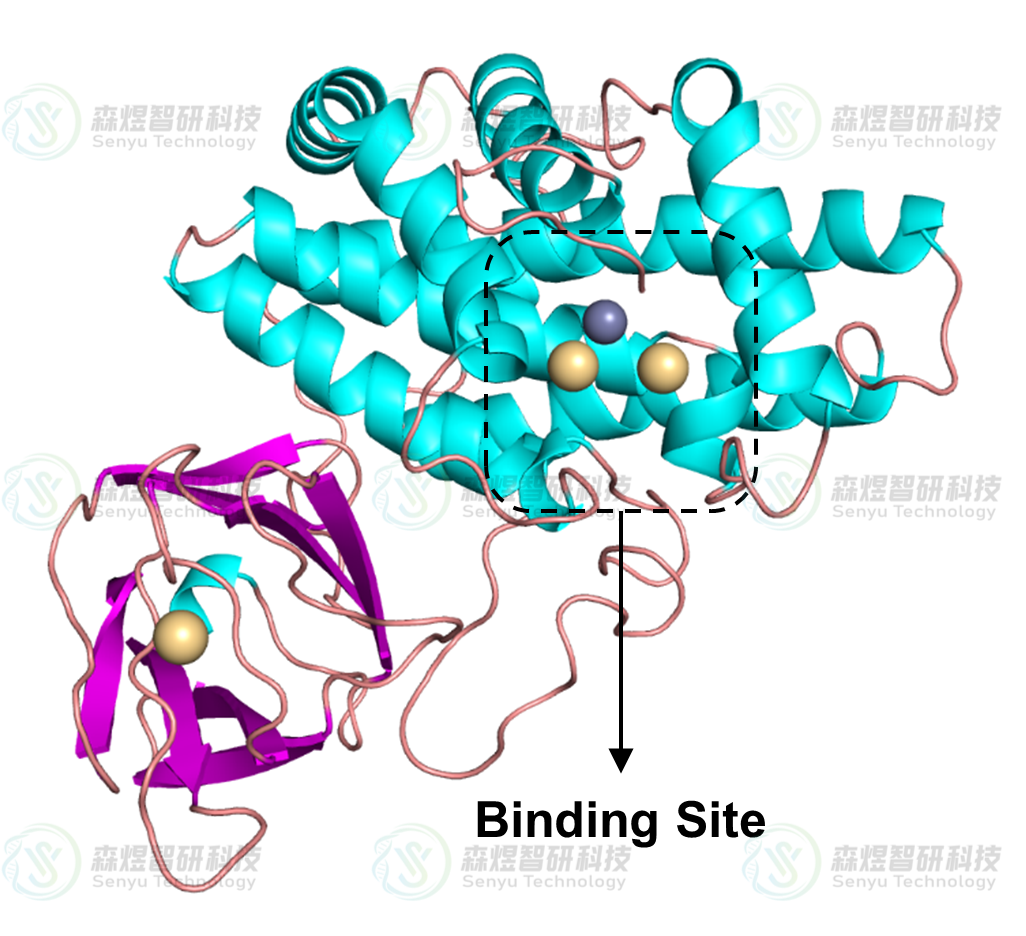
图4 CPA蛋白三维结构示意图,黑色虚线表示底物和蛋白结合位点。
分子对接的结果在空间结构上可能存在不合理的原子接触,可以采用能量优化的方法对这些作用力进行释放,使其更趋于稳定结构。能量优化采用Amber14力场[7],优化过程分两步进行:先进行1000步的最陡下降法(Steepest Descent Method)优化,再用500步的共轭梯度法(Conjugate Gradient Method)对结构进行进一步优化,并将最终的结果作为后续分析的模型,最终得到的复合物结构如图5所示。
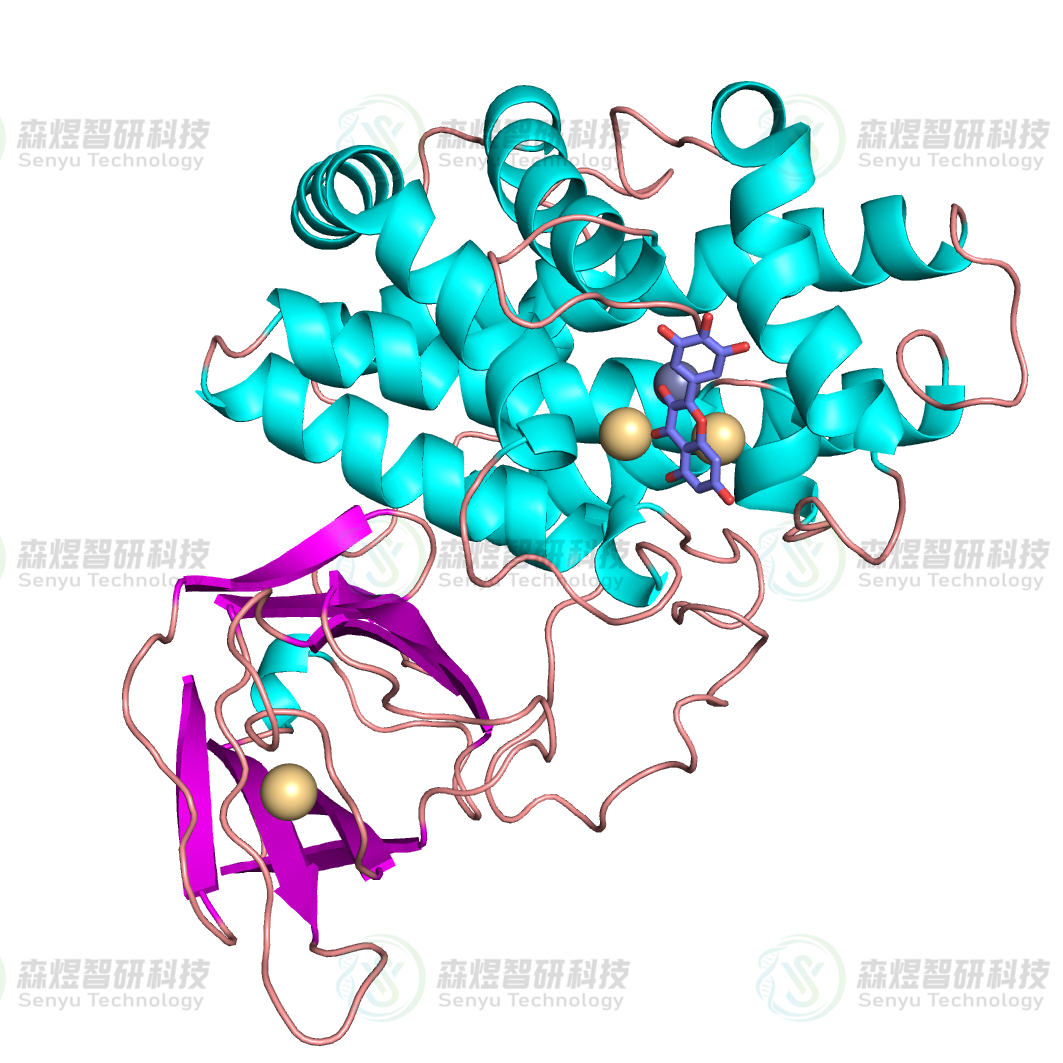
图5 CPA蛋白与MYR分子对接得到的复合物结构
MD模拟采用Gromacs 2018.4程序[8],在恒温恒压以及周期性边界条件下进行。应用Amber14SB全原子力场[9],TIP3P水模型[10]。在MD模拟过程中,所有涉及氢原子的键采用LINCS算法[11]进行约束,积分步长为2 fs。静电相互作用采用(Particle-mesh Ewald)PME方法[12]计算。非键相互作用截断值设为10 Å,每10步更新一次。采用V-rescale [13]温度耦合方法控制模拟温度为298.15 K,采用Parrinello-Rahman方法[14]控制压力为1 bar。首先,采用最陡下降法对两个体系进行能量最小化,以消除原子间过近的接触;然后,在298.15 K条件下分别进行1 ns的NVT和NPT平衡模拟;最后,对体系进行50 ns的MD模拟,每10 ps保存一次构象,模拟结果可视化采用Gromacs内嵌程序和VMD完成。
均方根偏差(Root mean square deviation, RMSD)表示某一时刻的构象与目标构象所有原子偏差的加和,是衡量体系是否稳定的重要依据。本文将CPA蛋白与小分子MYR对接后形成的复合物体系命名为Complex(后面都以此缩写代替)。图6给出了Complex复合物中所有原子的RMSD随时间的变化,Complex整体的RMSD在模拟初期经历一个明显的上升,可能是此时复合物体系中蛋白刚与溶剂接触,与溶剂间相互作用较强,使其波动明显。随后在20 ns左右,此时体系的RMSD开始趋于稳定,稳定后的平均值为0.291±0.028 nm,表明基于该力场参数下动力学模拟是稳定可靠的,可用于后续的进一步深入分析。

图6 CPA蛋白与MYR复合物RMSD值随模拟时间的变化
均方根涨落(Root mean square fluctuation, RMSF)可以表示蛋白质氨基酸残基的柔性大小,图7分析了整个体系中所有氨基酸的RMSF值。结果显示在Complex体系中CPA蛋白整体结构的RMSF值都较小,仅有S107-P119结构域的RMSF波动较大,表明整个CPA蛋白在模拟过程中结构并未发生明显解旋或变构。如图,对S107-P119结构域分析发现该区域是蛋白结构中一段柔性较大的Loop区,并且该段结构域离底物结合位点较远,不会对小分子化合物与蛋白间结合产生影响。整个CPA蛋白的RMSF整体波动值整体较小,并未因为MYR小分子的结合发生大幅波动,表明小分子与蛋白的结合是稳定的。
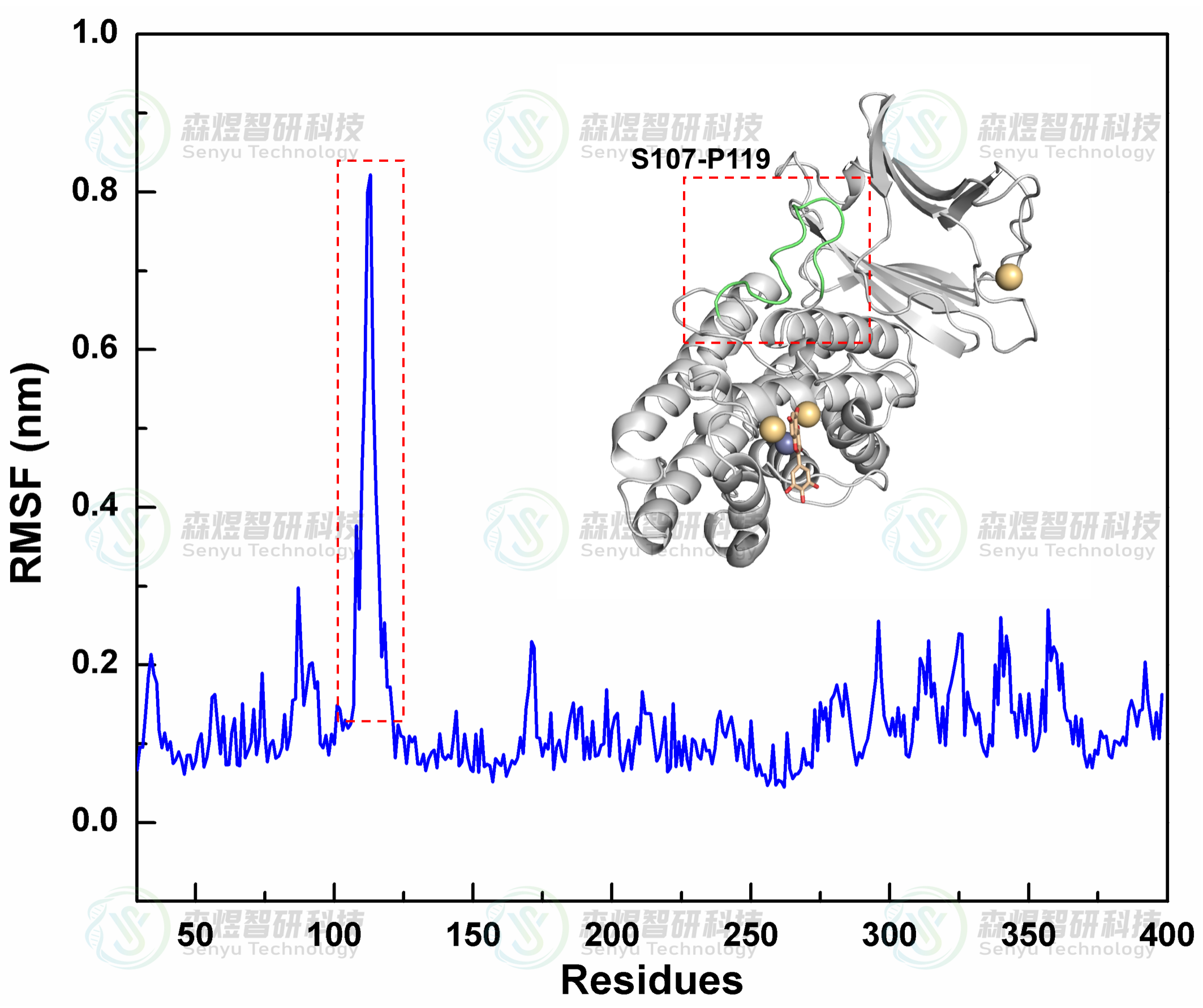
图7复合物体系中CPA蛋白氨基酸的RMSF分布
溶剂可及表面积(SASA)可以通过表征蛋白体系在溶剂环境中可被溶剂接触的分子表面积来分析模拟过程中蛋白在溶剂条件下的结构体积变化,SASA值越大表明蛋白结构更松散,体积也更大。为了研究复合物体系在模拟过程中的结构变化情况,本文对Complex复合物体系的SASA进行了分析,结果如图8所示。整个体系的SASA值在整个模拟过程中并未因底物的结合出现明显大幅波动,表明蛋白和MYR分子形成的复合物在溶剂体系中是非常稳定的,并未因小分子的结合导致整体蛋白结构出现明显的构象变化。
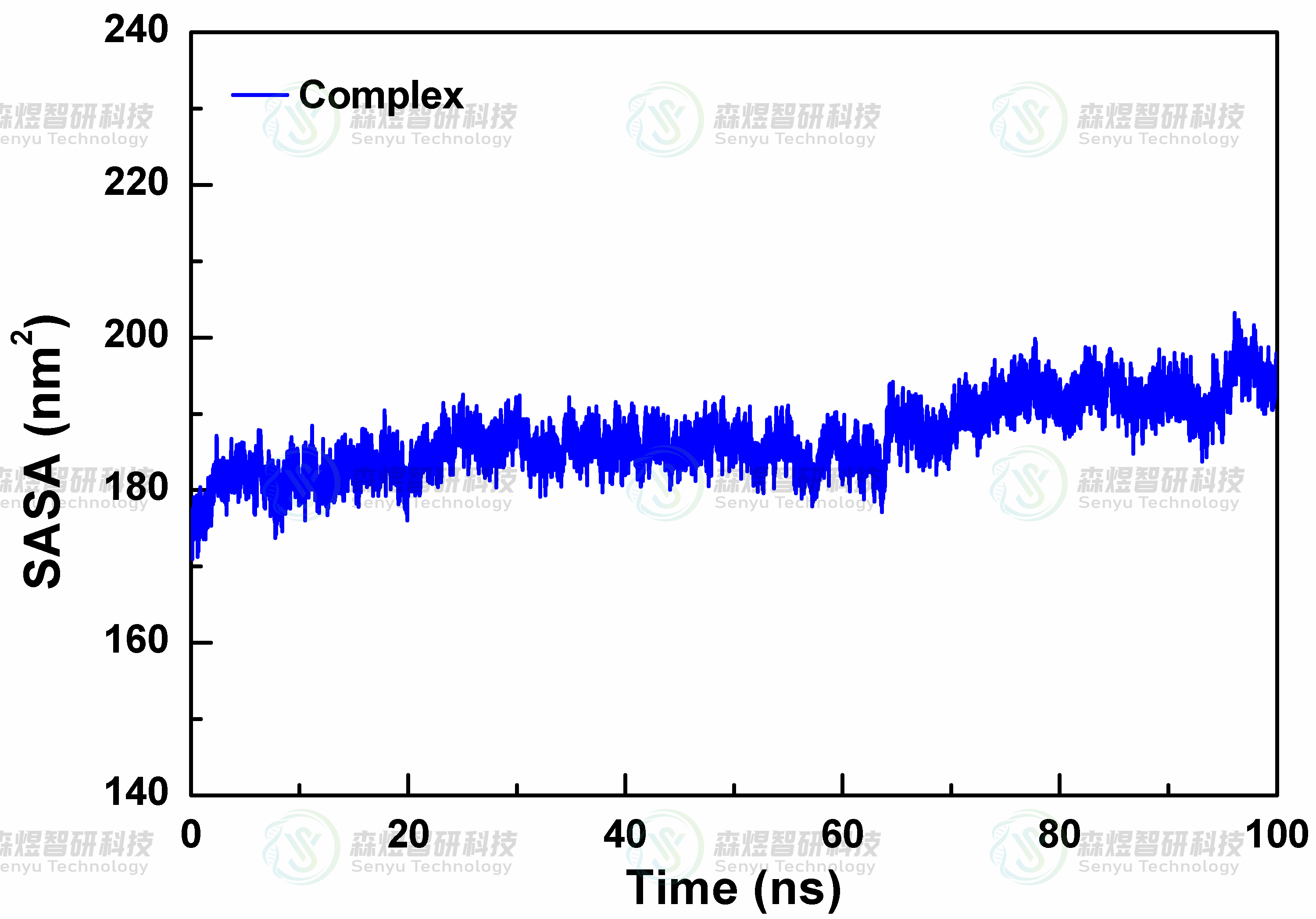
图8复合物体系SASA值随模拟时间的变化
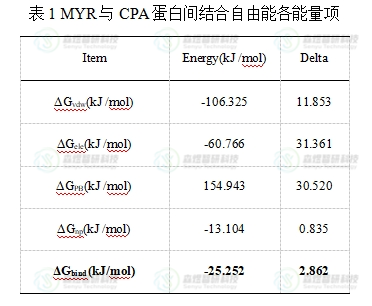
接下来,本文借助MM/PBSA方法计算了RMSD较为稳定的80-100 ns区间时CPA蛋白与MYR分子底物间的结合自由能,结果如表1所示,CPA与MYR间总的结合自由能为 -25.252 kcal/mol,其中体系的静电相互作用包括真空条件下的静电相互作用和极性溶剂化能(ΔGele+ΔGPB)为94.177 kcal/mol,对小分子和蛋白间的结合无明显促进作用;非极性作用可以作为疏水相互作用能,包括范德华力作用和非极性溶剂化自由能(ΔGvdw+ΔGnp),非极性相互作用为-119.429 kcal/mol,因此可以推断CPA蛋白与MYR分子的结合过程中,疏水相互作用能是主要的驱动力。
随后,为了进一步确认CPA蛋白与底物分子主要相互作用区域及哪些关键氨基酸参与了化合物与蛋白间的相互作用,本文提取MD模拟之后的复合物构象,对蛋白间的相互作用模式做了分析,结果如图9所示。从图9a和c中可以看出,MYR分子主要结合在CPA蛋白中Zn2+和Cd2+离子上方的疏水性空腔中,分子上整个占据了催化底物结合的空腔。从图9b和d中可以看出,MYR分子主要结合在由Trp29、Asp30、Gly31、Tyr93、Val177和Thr181氨基酸及Zn2+和Cd2+离子组成的空腔中,其中MYR分子环上的羟基与周围的Trp29、Asp30、Gly31、Val177和Thr181氨基酸间形成了较多的氢键作用,与Tyr93氨基酸间形成了疏水相互作用,同时MYR分子上的羰基O原子还与两个金属离子间产生了配位相互作用,进一步增强了MYR分子与CPA蛋白的亲和力。
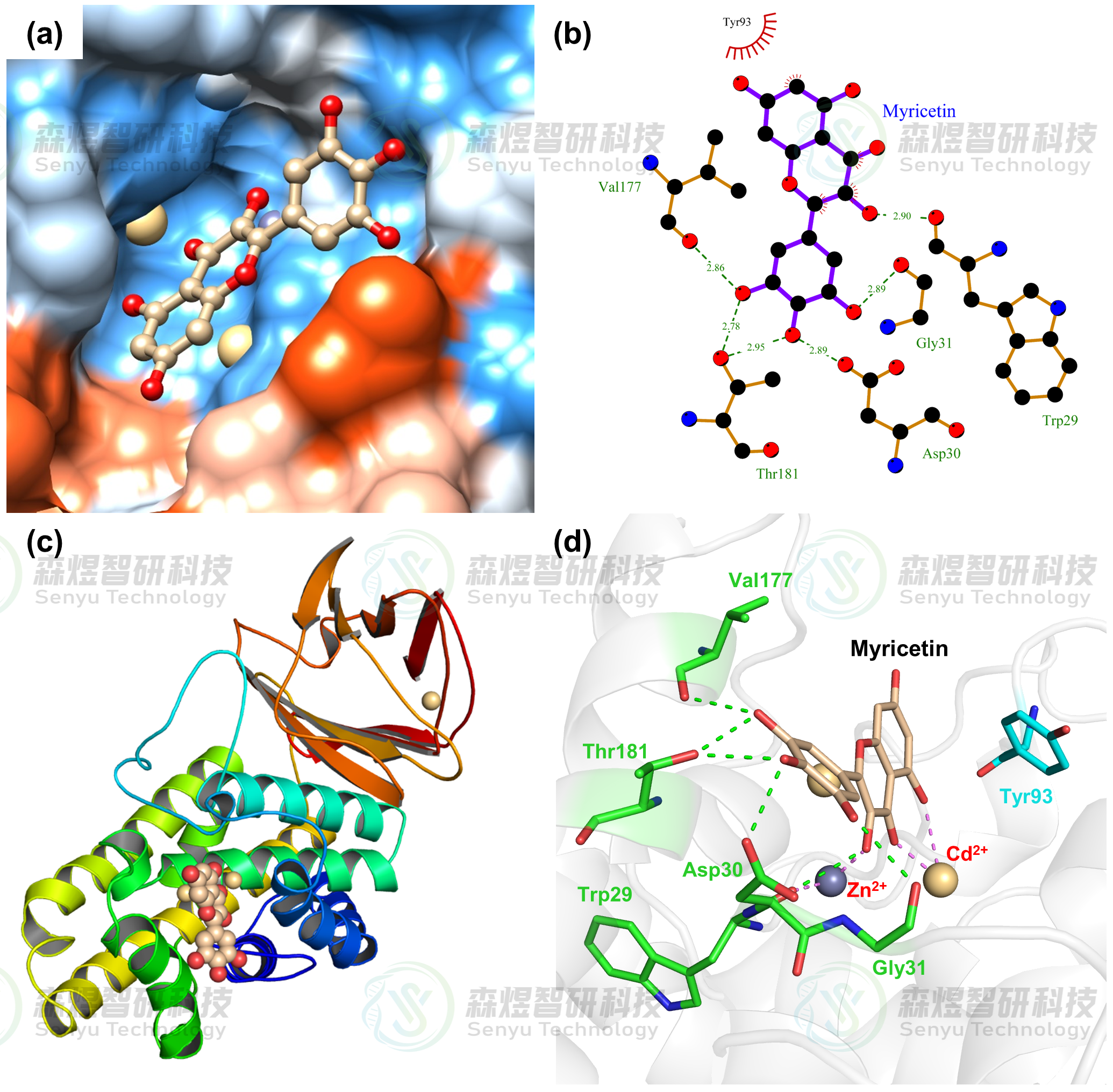
图9 MYR与CPA的结合模式图(a:MYR在CPA亲疏水性表面的结合,蓝色和橙色分别表示蛋白表面的疏水和亲水部分;b:MYR与CPA的二维结合模式,绿色虚线表示氢键作用,红色齿轮状表示疏水作用;c:MYR在CPA蛋白三维结构中的位置;d:MYR与CPA的三维结合模式图,绿色虚线表示氢键作用,紫色虚线表示配位作用)
本文结合同源建模、分子对接和MD模拟方法研究了MYR分子与CPA蛋白间结合的稳定性及相互作用模式,通过100ns动力学分析发现,MYR与CPA蛋白结合较为稳定,并未导致蛋白整体结构大幅波动。通过MM/PBSA方法计算得到的结合自由能结果表明MYR在CPA蛋白口袋中主要通过氢键作用和非极性的疏水相互作用促进其稳定结合并与占据底物结合口袋,阻止底物与蛋白间的结合催化作用。本研究结果为MYR化合物与CPA蛋白的分子识别以及能否稳定结合并竞争性抑制该酶活性研究提供了理论指导。
参考文献
[1] Sansen S , Yano J K , Reynald R L , et al. Adaptations for the Oxidation of Polycyclic Aromatic Hydrocarbons Exhibited by the Structure of Human P450 1A2[J]. Journal of Biological Chemistry, 2007, 282(19):14348-14355.
[2] Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., Lepore, R., Schwede, T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1), W296-W303 (2018).
[3] Laskowski R A, Macarthur M W, Moss D S, et al. PROCHECK: a program to check the stereochemical quality of protein structures[M]. 1993.
[4] Sanner M F. Python: a programming language for software integration and development. J. Mol. Graph. Model., 1999, 17(1): 57-61.
[5] Stewart J J. MOPAC: a semiempirical molecular orbital program. J. Comput. Aid. Mol. Des., 1990, 4(1): 1-105.
[6] Stewart J J P. Optimization of parameters for semiempirical methods. III. Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl, Pb, and Bi. John Wiley & Sons Inc., 1991.
[7] Maier J A, Martinez C, Kasavajhala K, et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 2015, 11(8): 3696-3713.
[8] Van d S D , Lindahl E , Hess B , et al. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 2005, 26(16):1701-1718.
[9] Maier J A, Martinez C, Kasavajhala K, et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 2015, 11(8): 3696-3713.
[10] Jorgensen W L , Chandrasekhar J , Madura J D , et al. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 1983, 79(2):926-935.
[11] Hess B , Bekker H , Berendsen H J C , et al. LINCS: A linear constraint solver for molecular simulations. Journal of Chemical Theory & Computation, 1997, 4(1):1463–1472.
[12] Darden T A , York D M , Pedersen L G . Particle mesh Ewald-an N.log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 1992, 98:10089-10092.
[13] Berendsen H J C , Postma J P M , Van Gunsteren W F , et al. Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 1984, 81(8):3684-3690.
[14] R Martonák, Laio A , Parrinello M . Predicting Crystal Structures: The Parrinello-Rahman Method Revisited. Physical Review Letters, 2003, 90(7):075503.
研究内容:本文采用分子动力学模拟方法研究了姜黄素和白藜芦醇与直链淀粉的分子识别
V型直链淀粉(Amylose)由35个葡萄糖单位组成,葡萄糖单位间以α-糖苷键相连。直链淀粉结构通过GLYCAM网站构建,姜黄素(Curcumin)和白藜芦醇(Resveratrol)结构通过PubChem下载。三个分子的结构如图1所示。
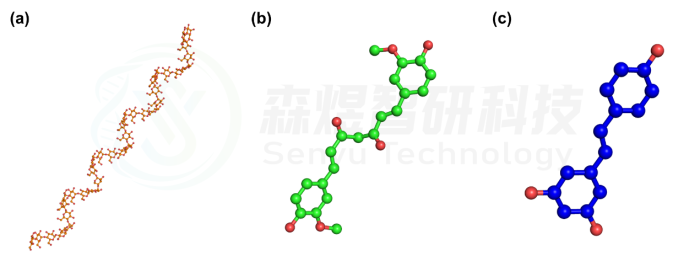
图1 Amylose三维结构(a);Curcumin三维结构(b);Resveratrol三维结构(c)
小分子与支链淀粉复合物分子动力学模拟结果
对Curcumin和Resveratrol与Amylose复合物分别进行MD模拟,以观察两个小分子与支链淀粉的结合是否稳定及其相互作用机制。将Amylose-Curcumin和Amylose-Resveratrol复合物体系进行MD模拟后,分别提取模拟后的最终构象,如图2所示,经过100 ns模拟,Amylose-Curcumin和Amylose-Resveratrol复合物体系中两个小分子依然能稳定与Amylose结合,只是在模拟过程中螺旋状的Amylose分子发生了一定的空间构象改变。
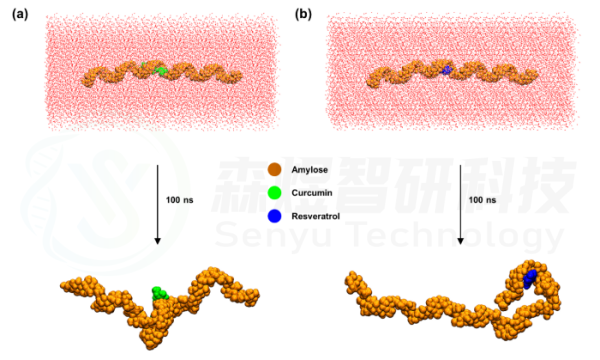
图2 Amylose-Curcumin和Amylose-Resveratrol复合物体系模拟前后空间构象
随后,为了更详细的观察两个小分子与支链淀粉复合物在整个模拟过程中的动态变化过程,我们对两个体系在模拟过程中复合物的构象进行了分析,每间隔20 ns截取一个稳定构象,Amylose-Curcumin结果如图3所示。在整个模拟过程中,尽管Amylose分子的空间构象有所改变,但是整个过程中Curcumin分子都与Amylose分子稳定的结合。
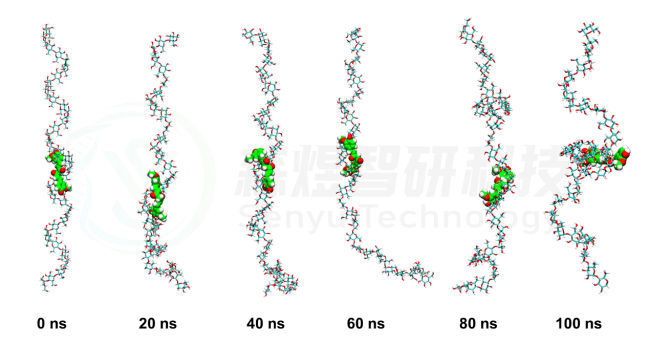
图3 Amylose-Curcumin复合物的构象随模拟时间的变化
Amylose-Resveratrol结果如图4所示。在整个模拟过程中,尽管Amylose分子的空间构象有所改变,但是整个过程中Resveratrol分子都与Amylose分子稳定的结合。
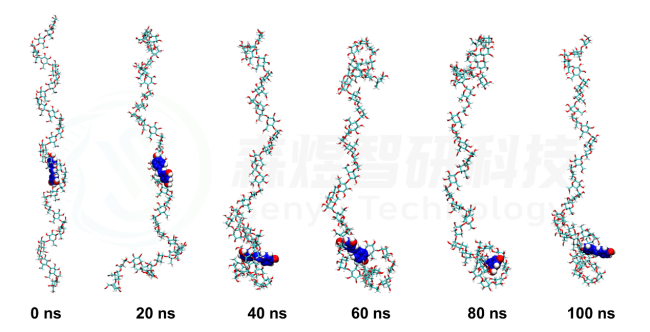
图4 Amylose-Resveratrol复合物的构象随模拟时间的变化
均方根偏差(Root mean square deviation, RMSD)表示某一时刻的构象与目标构象所有原子偏差的加和,是衡量体系是否稳定的重要依据。图3a给出了两个复合体系中所有原子的RMSD值随模拟时间的变化,可以观察到两个体系的RMSD值在模拟过程中都存在一定幅度的波动,可能是因为直链淀粉分子两端在溶剂分子的影响下发生了一定的构象改变。两个体系的RMSD平均值分别为1.352±0.476 nm和1.638±0.308 nm,两个体系RMSD的波动幅度分别为35.21%和18.80%,两个体系整体的波动幅度都挺大,但如此大的波动幅度是否影响两个小分子与Amylose间的相互作用需进一步深入分析。
回转半径(Radius of Gyration, Rg)可以用来衡量分子整体结构的密实度,如果分子的空间结构很稳定,其Rg将保持一个相对稳定的值,Rg变化越大表明体系越膨胀。图3b给出了两个复合体系中所有原子的Rg值随模拟时间的变化,模拟初期,两个体系的Rg值小幅降低,随后在60 ns之前都趋于一个较为稳定状态,随后在70 ns左右,两个体系的Rg值都发生了一定程度的下降,此时两个体系中的Amylose分子构象发生了改变,结合前面的模拟前后构象对比分析,表明在模拟后期两个体系中的Amylose分子构象发生了折叠,但两个小分子依然稳定的结合在Amylose分子上。两个体系整个过程中Rg的平均值分别为2.828±0.445 nm和2.764±0.296 nm。
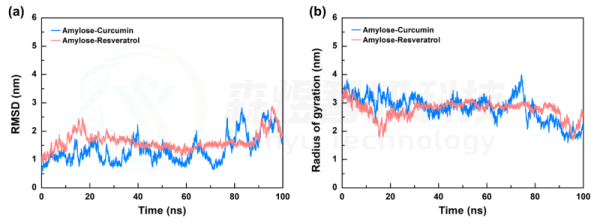
图5 Amylose-Curcumin和Amylose-Resveratrol复合物体系的RMSD和Rg值随模拟时间的变化
动力学模拟中复合物密度分析
随后,为了进一步分析两个复合物在模拟过程中的结合紧密程度,分别对两个体系中复合物,Amylose和两个小分子的密度沿盒子 z 轴的密度分布进行分析。从图4中可以看出,两个小分子在模拟过程中都是结合在Amylose分子中间的。从Amylose分子的密度分布可以发现,在Amylose-Curcumin体系中,Amylose分子最大密度更大,表明该体系中Amylose分子折叠得更厉害,同时Curcumin稳定结合在该位置;而在Amylose-Resveratrol体系中,mylose分子最大密度更小,表明该体系中Amylose分子结构更舒展,同时Resveratrol分子结合在密度最大位置旁边,单整个过程中Resveratrol也是一直稳定结合在Amylose分子上的。
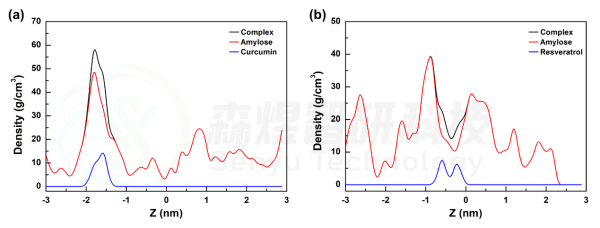
图6 Amylose-Curcumin(a)和Amylose-Resveratrol(b)复合物体系中各组分的密度分布
复合物与溶剂间氢键分析
当复合物置于溶剂盒子中时,其与溶剂分子间的氢键变化可以表征体系中复合物内小分子与Amylose分子间结合是否稳定,若Amylose分子空间构象发生明显折叠聚集,则其与溶剂水分子间的氢键会明显减少,反之则维持稳定;同样若小分子在模拟过程中从Amylose分子上游离,那小分子与溶剂分子间的氢键数会增加。两个体系中复合物、Amylose分子和小分子与溶剂水分子间的氢键数量随模拟时间的变化如图5所示。结果显示,在Amylose-Curcumin体系中,Amylose分子与水分子间的氢键较模拟初始时有一定减少,表明该体系中Amylose分子经过一定时间模拟后螺旋结构发生了变构折叠,而Curcumin分子与水分子间的氢键一直维持稳定,表明小分子与Amylose分子间一直稳定结合,并未出现游离。在Amylose-Resveratrol体系中,Amylose分子与水分子间的氢键在模拟过程中有一定减少,随后又增长回来,表明该体系中Amylose分子在模拟过程中发生过一定程度的变构折叠,但随着模拟的进行又舒展开来,Resveratrol分子与水分子间的氢键一直维持稳定,表明小分子与Amylose分子间一直稳定结合,并未出现游离。
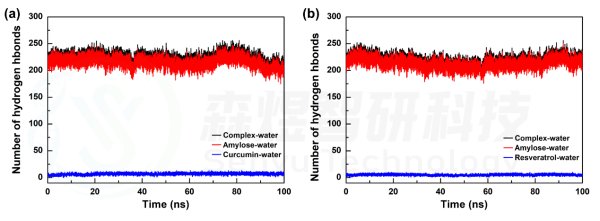
图7 Amylose-Curcumin(a)和Amylose-Resveratrol(b)复合物体系中各组分与水分子间的氢键随模拟时间的变化
分子间氢键和疏水作用分析
促进分子间结合的弱相互作用主要包括氢键作用、静电作用、疏水作用、π-π、π-sigma 和π-阳离子共轭作用,通常两个分子间的结合主要通过氢键作用和疏水相互作用作用驱动力。因此,本文对两个体系中Curcumin和Resveratrol分子与Amylose间的氢键作用和疏水作用随模拟时间的变化进行了分析,结果如图6所示。从图中可以发现,Curcumin分子与Amylose分子间的氢键作用较Resveratrol分子略强,两个体系中整个模拟过程中的氢键平均值分别为1.22和0.79。图6b为两个体系中Curcumin和Resveratrol分子与Amylose间疏水相互作用随模拟时间的变化,两个体系的平均值分别为2.46和1.99。两个体系中Curcumin分子与Amylose分子间的氢键作用的疏水作用都略强于Resveratrol分子,表明可能Curcumin分子与Amylose分子间的相互作用更强。
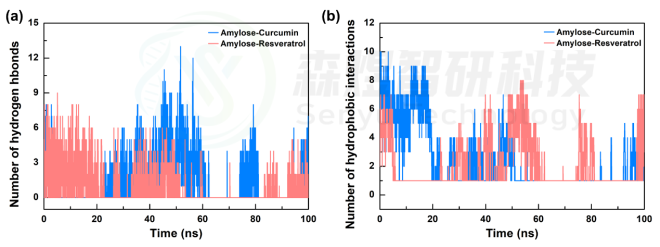
图8 Amylose-Curcumin和Amylose-Resveratrol复合物体系中小分子与Amylose间的氢键作用(a)和疏水作用(b)随模拟时间的变化
复合物SASA分析
溶剂可及表面积(Solvent accessible surface area, SASA)可以通过表征复合物体系在溶剂环境中可被溶剂接触的分子表面积来分析模拟过程中小分子和Amylose分子在溶剂条件下的结构体积变化,SASA值越大表明结构更松散,体积也更大,反之。在Amylose-Curcumin复合物体系中,Amylose分子的SASA值较初始状态时SASA值降低了约13%,表明该体系中Amylose分子经过一定时间模拟后螺旋结构发生了变构折叠,整体体积减小;而在Amylose-Resveratrol复合物体系中,虽然模拟过程中Amylose分子的SASA值有所下降,但末态相较于初态SASA值几乎没有下降,表明该体系中Amylose分子在模拟过程中发生过一定程度的变构折叠,但随着模拟的进行又舒展开来,最终Amylose分子体积无明显变化。进一步分析了两个体系中的小分子的SASA值,可以发现两个小分子的SASA值无明显减小,表明整个模拟过程中小分子的空间构象并未发生明显变化。从图7b中两个复合物的SASA值变化也可以看出,在Amylose-Curcumin复合物体系中,由于Amylose分子的变构折叠使整个复合物体系的SASA略有减小,而在Amylose-Resveratrol复合物体系中无明显变化。上述结果进一步表明,在Amylose-Curcumin复合物体系Amylose分子变构折叠,体积减小,同时Curcumin分子结合在Amylose分子变构折叠区域,可能进一步增强两个分子间的相互作用,而Amylose-Resveratrol复合物体系中Amylose分子仅发生小幅变化,并且Resveratrol分子也并未结合在该位置,因此它们间的相互作用可能与初始状态无明显变化。
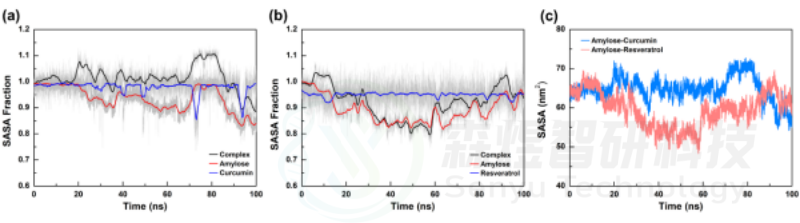
图9 Amylose-Curcumin和Amylose-Resveratrol复合物体系中SASA随模拟时间的变化(a:各组分的SASA比例随模拟时间的变化;b:两个复合物体系SASA值随模拟时间变化)
相互作用模式分析
图8给出了Curcumin分子和Resveratrol分子与Amylose分子间的结合模式,从图8a和b中可以看出,Curcumin分子结合在Amylose分子中17-25位葡萄糖分子形成的螺旋空腔中,Curcumin分子分别与17位葡萄糖分子上的O6、20位葡萄糖分子上的O6和25位葡萄糖分子上的O3间形成氢键作用,同时还与18、19、21、22和23位葡萄糖基间形成疏水相互作用,进一步促进两个分子间稳定结合。从图8b和c中可以看出,Resveratrol分子结合在Amylose分子中19-24位葡萄糖分子形成的螺旋空腔中,Resveratrol分子分别与22位葡萄糖分子上的O6和24位葡萄糖分子上的O2间形成氢键作用,同时还与19、20、21和23位葡萄糖基间形成疏水相互作用,进一步促进两个分子间稳定结合。
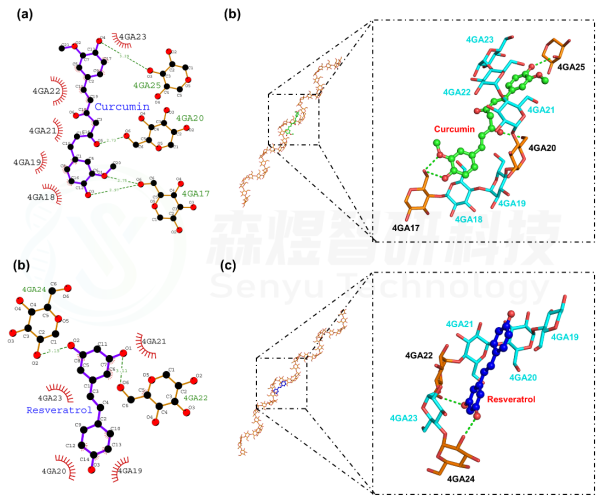
图10 Curcumin分子和Resveratrol分子与Amylose分子间的结合模式(a:Curcumin分子与Amylose分子间的2D结合模式,齿状糖基分子为疏水相互作用,绿色虚线为氢键作用;b:Curcumin分子与Amylose分子间的3D结合模式,Cyan色糖基为疏水基团,绿色虚线为氢键作用;c:Resveratrol分子与Amylose分子间的2D结合模式,齿状糖基分子为疏水相互作用,绿色虚线为氢键作用;d:Resveratrol分子与Amylose分子间的3D结合模式,Cyan色糖基为疏水基团,绿色虚线为氢键作用)
本文采用MD模拟方法对Curcumin分子和Resveratrol分子与Amylose分子间的分子识别及其稳定性进行了对比分析,通过对比分析发现,两个小分子都能与Amylose分子间产生稳定的相互作用。进一步对比两个分子与Amylose分子间的氢键作用和疏水相互作用结果表明Curcumin分子与Amylose分子间的相互作用可能较Resveratrol分子更强。
参考文献
[1] Sanner M F. Python: a programming language for software integration and development. J. Mol. Graph. Model., 1999, 17(1): 57-61.
[2] Stewart J J. MOPAC: a semiempirical molecular orbital program. J. Comput. Aid. Mol. Des., 1990, 4(1): 1-105.
[3] Stewart J J P. Optimization of parameters for semiempirical methods. III. Extension of PM3 to Be, Mg, Zn, Ga, Ge, As, Se, Cd, In, Sn, Sb, Te, Hg, Tl, Pb, and Bi. John Wiley & Sons Inc., 1991.
[4] Maier J A, Martinez C, Kasavajhala K, et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. Journal of Chemical Theory and Computation, 2015, 11(8): 3696-3713.
[5] Van d S D , Lindahl E , Hess B , et al. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 2005, 26(16):1701-1718.
[6] J. Comput. Chem. GLYCAM06: A generalizable biomolecular force field. Carbohydrates[J]. 2008.
[7] Jorgensen W L , Chandrasekhar J , Madura J D , et al. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 1983, 79(2):926-935.
[8] Hess B , Bekker H , Berendsen H J C , et al. LINCS: A linear constraint solver for molecular simulations. Journal of Chemical Theory & Computation, 1997, 4(1):1463–1472.
[9] Darden T A , York D M , Pedersen L G . Particle mesh Ewald-an N.log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 1992, 98:10089-10092.
[10] Berendsen H J C , Postma J P M , Van Gunsteren W F , et al. Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 1984, 81(8):3684-3690.
[11] R Martonák, Laio A , Parrinello M . Predicting Crystal Structures: The Parrinello-Rahman Method Revisited. Physical Review Letters, 2003, 90(7):075503.
研究内容:本文采用分子动力学模拟方法研究了不同小分子自组装的结构差异。
实验研究发现,黄芩苷与血根碱按照1:1的比例放入水溶剂环境中能自组装形成纳米团簇;β-甘草次酸自身在比例为DMSO:H2O的溶剂环境中也能自组装形成纳米团簇。为了研究它们最终自组装形成的结构特征以及整个自组装过程中分子间的主要驱动力,本文采用分子动力学(Molecular dynamics, MD)模拟方法对两个体系中小分子的自组装过程展开了研究。
首先构建6 nm×6 nm×6 nm的模拟盒子,并分别随机填充20个黄芩苷、20个血根碱和40个β-甘草次酸分子,组成2个模拟体系。MD模拟采用Gromacs 2018.4程序[1],在恒温恒压以及周期性边界条件下进行。应用GAFF全原子力场,TIP3P水模型[2]。在MD模拟过程中,所有涉及氢键采用LINCS算法[3]进行约束,积分步长为2 fs。静电相互作用采用(Particle-mesh Ewald)PME方法[4]计算。非键相互作用截断值设为10 Å,每10步更新一次。采用V-rescale [5]温度耦合方法控制模拟温度为298.15 K,采用Parrinello-Rahman方法[6]控制压力为1 bar。首先,采用最陡下降法对四个体系进行能量最小化,以消除原子间过近的接触;然后,在298.15 K进行100 ps的NVT平衡模拟;最后,分别对2个不同体系分别进行100 ns的MD模拟,每10 ps保存一次构象,模拟结果可视化采用Gromacs内嵌程序和VMD完成。
模拟收敛参数分析
均方根偏差(Root mean square deviation, RMSD)表示某一时刻的构象与目标构象所有原子偏差的加和,是衡量体系是否稳定的重要依据。图1a和b分别给出了Sanguinarine-Baicalin和Glycyrrhetinic acid 2个体系中小分子所有原子的RMSD值随模拟时间的变化。从图1a中可以看出,Sanguinarine-Baicalin体系的RMSD值在模拟过程中的波动相对较小,而Glycyrrhetinic acid的RMSD波动较大,二者在整个模拟过程中的平均值分别为3.024±0.103 nm和3.051±0.144 nm。整体来讲,Sanguinarine-Baicalin体系在模拟过程中RMSD的波动相对较小,表明这个体系在模拟过程中更加稳定。

图1 2个体系所有原子的RMSD(a和b)随模拟时间的变化
随着纳米粒子在自组装的进行,暴露在溶剂环境中的面积会逐渐减小,因此溶剂可及表面积(SASA)可以用来评价纳米粒子的紧密程度。图2a和b分别给出了2个体系在模拟过程中的溶剂可及表面积的相对变化,从整体上看,2个体系的SASA值在模拟过程中均出现了不同程度的降低,表明2个体系在模拟过程中均出现了不同程度的聚集现象。具体来讲,从图2中可以看出,2个体系的SASA值在模拟初始阶段都出现了非常明显的降低,随后减小速度放缓,并在40 ns后趋于稳定,2个体系的SASA平均值分别为69.940±4.864 nm2和92.328±4.126 nm2。由于两个体系中包含的分子不同,结构体积存在差异,不能横向对比,但两个体系中的SASA都明显降低并趋于稳定表明两个体系中的分子都能自组装形成更为紧密的纳米团簇,使其暴露在溶剂中的原子减少。
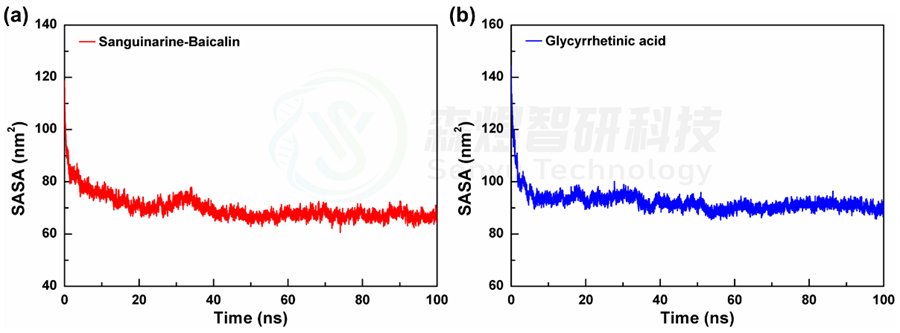
图2 2个体系溶剂可及表面积(a和b)随模拟时间的变化
聚类分析
为了研究模拟2个体系在模拟前后的构象变化以及自组装的结构紧密程度,本文采用Gromacs的cluster模块对四个体系中70-100 ns的模拟轨迹进行聚类分析,设置均方根偏差(Root mean square deviation, RMSD)截断值为1.2 nm。分别取体系最大簇的结构作为MD模拟的最终构象,如图3和4所示。从图3中可以看出,Sanguinarine-Baicalin体系自组装形成纤维状的纳米团簇结构。
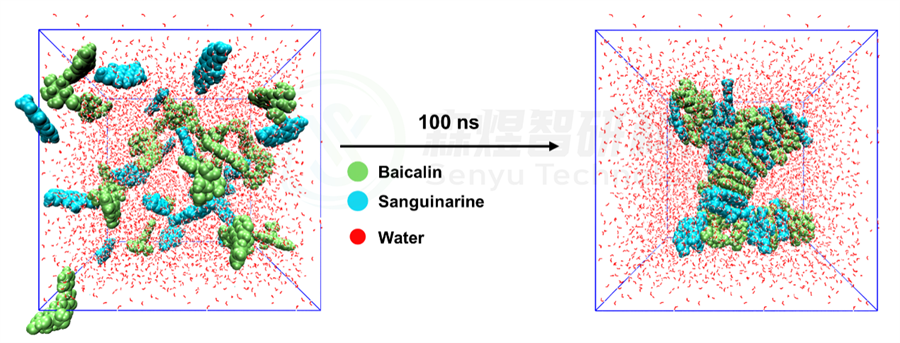
图3 Sanguinarine-Baicalin体系在模拟过程中的初末态结构变化
从图4中可以看出,经过100 ns的MD模拟之后,Glycyrrhetinic acid体系中所有小分子形成了比较稳定的,致密的类似球状的纳米团簇结构。
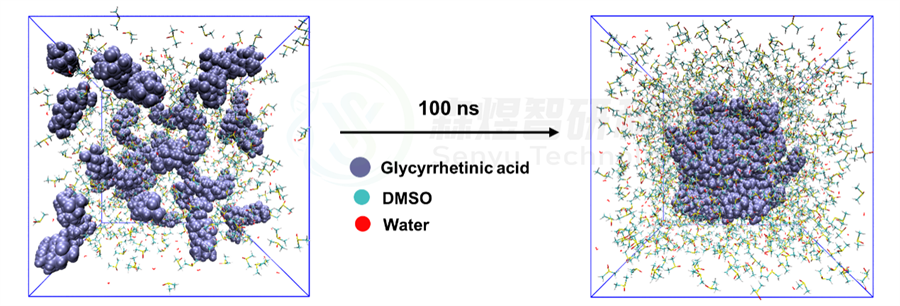
图4 Glycyrrhetinic acid体系在模拟过程中的初末态结构变化
随后,为了能更清楚的展现两个体系中小分子随着模拟进行不断自组装的动态过程,本研究对两个体系中的分子结构每间隔20 ns取一帧结构作为代表性构象进行绘图,如图6和图7所示,两个体系中的分子在分子间的相互作用下随着模拟的进行不短的聚集,最终形成具有一定形态的稳定纳米团簇结构。

图5 Sanguinarine-Baicalin体系在模拟过程中的结构随时间的变化

图6 Glycyrrhetinic acid体系在模拟过程中的结构随时间的变化
分子间相互作用分析
为了进一步研究体系自组装成纳米粒子过程中的驱动力,本文对MD模拟过程中的分子间的氢键数量进行了统计分析。如图7所示,从图中可以看出,在Glycyrrhetinic acid体系中,氢键作用相对较少,平均值为0.864±0.929个,而Sanguinarine-Baicalin体系中的氢键作用数量平均值为5.041±1.893个。整体来讲,Sanguinarine-Baicalin体系中的氢键数量比Glycyrrhetinic acid体系中明显更多,表明体系中分子间的相互作用更强,较多的氢键作用也可以使其组装的纳米粒子结构更加紧密。
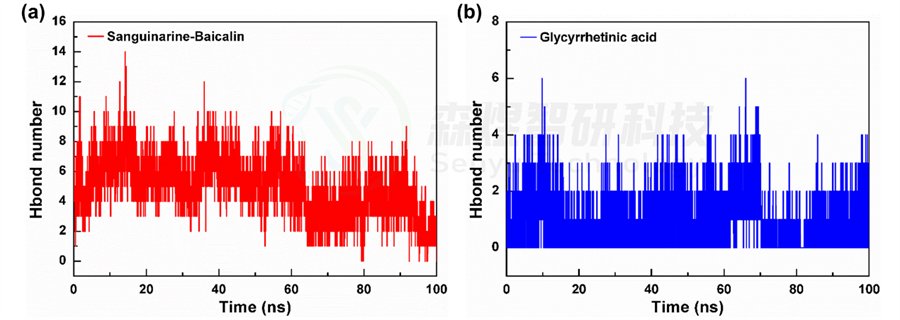
图7 Sanguinarine-Baicalin(a)和Glycyrrhetinic acid(b)体系中分子间的氢键数目随模拟时间的变化
随后,为了能更好的展现两个体系中分子间自组装的驱动力, 本文利用gmx_energy插件对两个体系中代表性分子间的相互作用能随模拟时间的变化进行了分析,结果如图8所示。其中Coul-SR代表静电能,主要作用类型为氢键或离子键等;LJ-SR代表范德华能,主要作用类型为各种共轭作用。在Sanguinarine-Baicalin体系中Sanguinarine和Baicalin分子间的Coul-SR平均值为-13.437±6.981 kJ/mol,LJ-SR平均值为-71.624±10.169 kJ/mol;在Glycyrrhetinic acid体系中,Glycyrrhetinic acid分子间的Coul-SR平均值为-2.350±6.798 kJ/mol,LJ-SR平均值为-64.286±8.683 kJ/mol。对比两个体系中自组装分子间的结合能可以发现,在Sanguinarine-Baicalin体系中Sanguinarine和Baicalin分子间的相互作用强于Glycyrrhetinic acid体系中Glycyrrhetinic acid分子间的相互作用,更强的相互作用能表明分子间更易自组装形成纳米团簇并且结构更加紧密。

图8 Sanguinarine-Baicalin(a)和Glycyrrhetinic acid(b)体系中分子间结合能随模拟时间的变化
分子间相互作用模式
通过前面分析不同分子体系的自组装过程中的相互作用能的强弱变化,发现Sanguinarine-Baicalin体系中分子间的相互作用力相对较强, Glycyrrhetinic acid体系中分子间的相互作用力相对较弱,因此为了深入研究不同结构所造成的的自组装差异,本文对MD模拟之后2个体系的构象进行了深入分析,如图9和图10所示。图9为Sanguinarine-Baicalin体系中分子间的相互作用示意图,从图9中可以看出,Sanguinarine分子和Baicalin分子主要通过分子中的大量的环状结构导致彼此间形成的Pi-Pi共轭作用促进其自组装,同时,Baicalin分子末端的羟基与Sanguinarine分子环上的氧原子间形成一组氢键作用,Baicalin分子内部的羟基与羰基氧原子间形成了分子内氢键作用(图9a)。Sanguinarine分子与Sanguinarine分子间主要是通过Pi-Pi共轭作用结合(图9b);Baicalin分子与Baicalin分子间促进其结合的主要有Pi-Pi共轭作用、分子间氢键和分子内氢键作用(图9c)。

图9 Sanguinarine-Baicalin体系中分子间的相互作用模式
图10为Glycyrrhetinic acid体系中分子间的相互作用示意图,从图10中可以看出,体系内的Glycyrrhetinic acid分子间主要通过分子中一个Glycyrrhetinic acid分子上的甲基与另一个分子上的六元环分子间形成的Pi-Sigma共轭作用促进其自组装,并且由于每个环上都连有甲基分子,导致分子间的空间位阻较大,Glycyrrhetinic acid分子之间没有形成明显的氢键作用。

图10 Glycyrrhetinic acid体系中分子间的相互作用模式
本文采用MD方法模拟了Sanguinarine-Baicalin和Glycyrrhetinic acid两个体系组装成纳米结构的过程进行了研究。通过分析分子间相互作用力的变化,表明两个体系中的分子都能组装形成较稳定的纳米结构,Sanguinarine-Baicalin体系中主要的驱动力包括Pi-Pi共轭作用、分子间氢键和分子内氢键作用;Glycyrrhetinic acid体系中主要的驱动力为Pi-Sigma共轭作用。对两个体系最终组装形成的纳米团簇结构分析发现,Sanguinarine-Baicalin体系最终共组装形成纤维状的纳米结构,Glycyrrhetinic acid体系最终自组装形成类似球状的纳米团簇结构。
参考文献
[1] Van d S D , Lindahl E , Hess B , et al. GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 2005, 26(16):1701-1718.
[2] Jorgensen W L , Chandrasekhar J , Madura J D , et al. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics, 1983, 79(2):926-935.
[3] Hess B , Bekker H , Berendsen H J C , et al. LINCS: A linear constraint solver for molecular simulations. Journal of Chemical Theory & Computation, 1997, 4(1):1463–1472.
[4] Darden T A , York D M , Pedersen L G . Particle mesh Ewald-an N.log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 1992, 98:10089-10092.
[5] Berendsen H J C , Postma J P M , Van Gunsteren W F , et al. Molecular dynamics with coupling to an external bath. Journal of Chemical Physics, 1984, 81(8):3684-3690.
[6] R Martonák, Laio A , Parrinello M . Predicting Crystal Structures: The Parrinello-Rahman Method Revisited. Physical Review Letters, 2003, 90(7):075503.

18817128943

扫码关注 森煜智研微信号

扫码关注 森煜智研公众号