


【写在前面】
本期推荐的是由南昌大学附属医院等研究团队合作近期发表于PeerJ 的一篇文章,揭示USP22可作为骨肉瘤糖酵解途径的关键调节因子。下面就先跟着小森一起来解读一下吧~
1. 本研究通过整合多种生物信息学工具并结合临床数据,系统、全面地探索了USP22在骨肉瘤发生、进展和预后中的功能,确定潜在机制。
2. 本研究通过多数据库挖掘治疗靶点,从GEO 数据库中获得与骨肉瘤相关的转录组学数据集,进而结合TCGA数据集、STRING数据库、DAVID数据库获得了344个共表达基因,提高了数据来源的广泛性。
3. 在利用生物信息学分析数据的基础上,结合体外实验,进一步验证USP22对骨肉瘤细胞生长的影响及其机制。“干湿结合”增强实验结果的可靠性。
总的来说,这篇文章研究了骨肉瘤糖酵解途径的关键调节因子USP22的致癌作用,通过整合生物信息学分析和体外实验分析并验证了USP22的下调可以抑制骨肉瘤细胞的糖酵解和生长,完整性高,逻辑思维也比较强!对于这个思路,小伙伴们有没有兴趣应用在自己的研究中呢?想要了解生信数据挖掘,或是对数据库和生信工具使用感到迷茫的老铁们,快来和小森聊聊,小森一定帮你有的放矢!
【题目及作者信息】

【摘要】(阅读原文为主)
骨肉瘤是最常见的原发性恶性骨肿瘤,但发病机制尚不清楚。研究表明,泛素特异性加工肽酶22(USP22)高表达,与癌症中的肿瘤恶性肿瘤和预后相关。作者团队整合了GEO和TCGA数据库的转录组学数据集和骨肉瘤的临床信息,通过差异表达分析、加权基因共表达网络分析以及富集分析,探讨了USP22共表达基因的生物学功能和信号通路。并体外实验证实了生物信息学分析的准确性和可靠性。结果表明,USP22在骨肉瘤的发生、发展和预后中起着关键作用。说明了USP22的下调可以抑制骨肉瘤细胞的糖酵解和生长。
【方法】
整合GEO和TCGA数据库的转录组学数据集和骨肉瘤的临床信息,以评估USP22在骨肉瘤中的表达和预后价值。进行差异表达分析和加权基因共表达网络分析(WGCNA)鉴定USP22相关共表达基因。通过基因本体论(GO)和京都基因与基因组百科全书(KEGG)富集分析,探讨USP22共表达基因的生物学功能和信号通路。为验证生物信息学分析的准确性,使用 siRNA 下调骨肉瘤细胞系 Sao-2中USP22 的表达,并评估其对细胞增殖、迁移、侵袭、凋亡和关键信号通路调控的影响。
【结果部分】
USP22在骨肉瘤中高表达,与预后不良相关
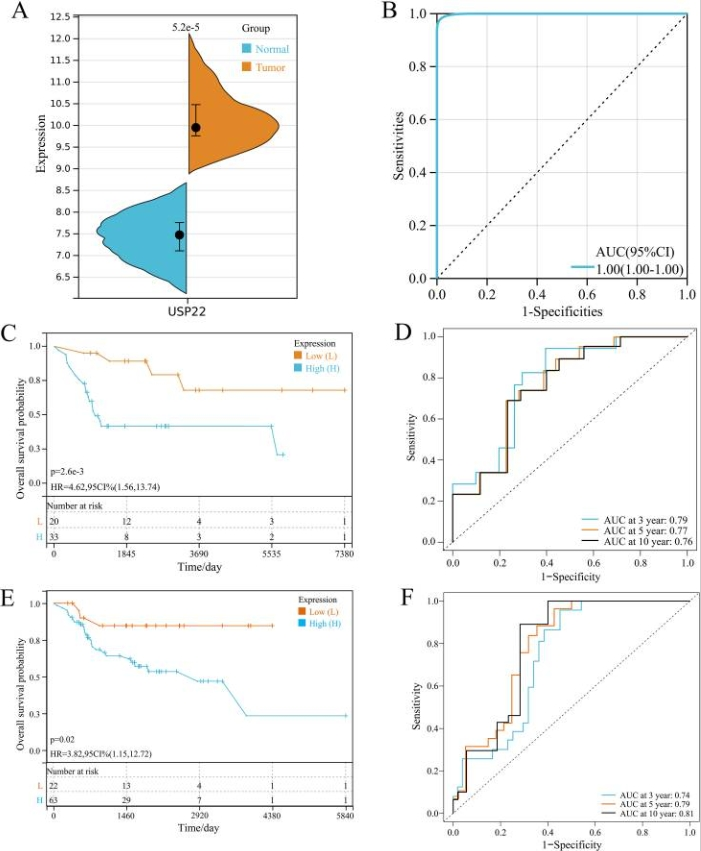
图1 USP22在骨肉瘤中的表达及预后分析。
USP22共表达基因的鉴定
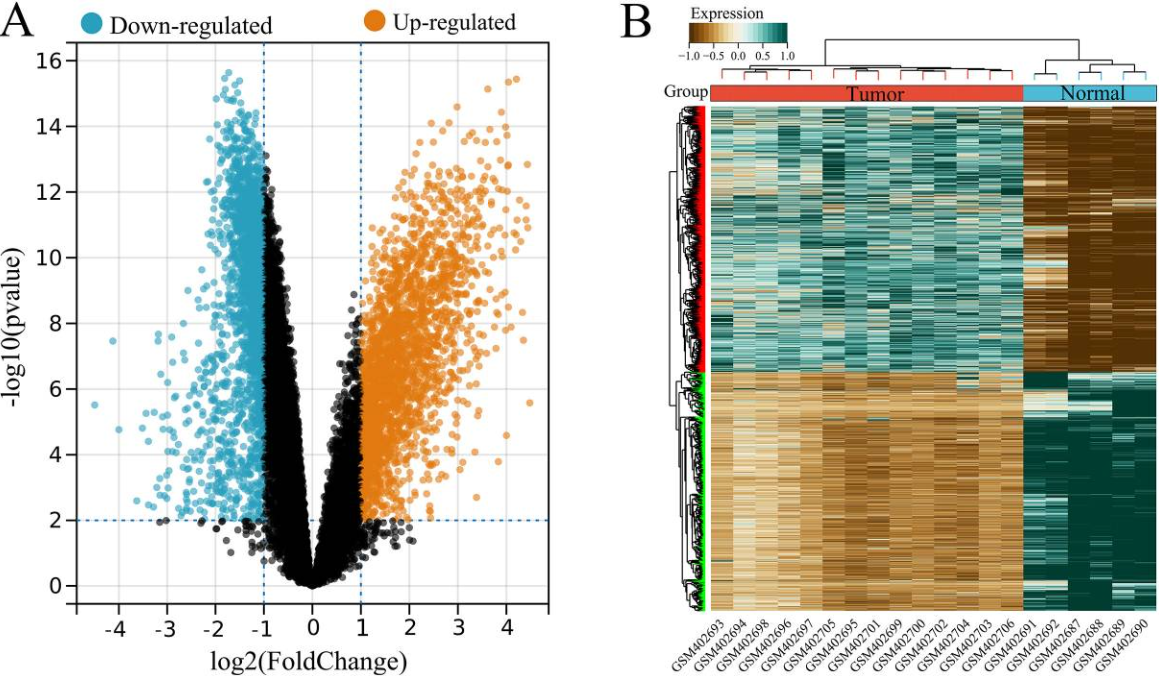
图2 GSE16088数据集中USP22相关差异表达基因 (DEGs) 的鉴定。
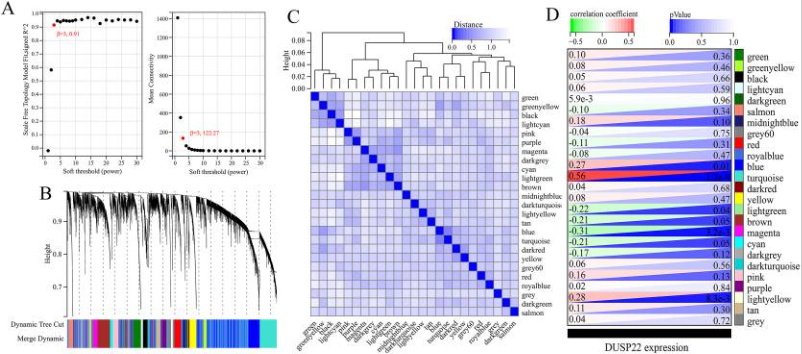
图3 使用加权基因共表达网络分析(WGCNA) 鉴定TCGA-OS数据集中USP22相关的共表达基因。
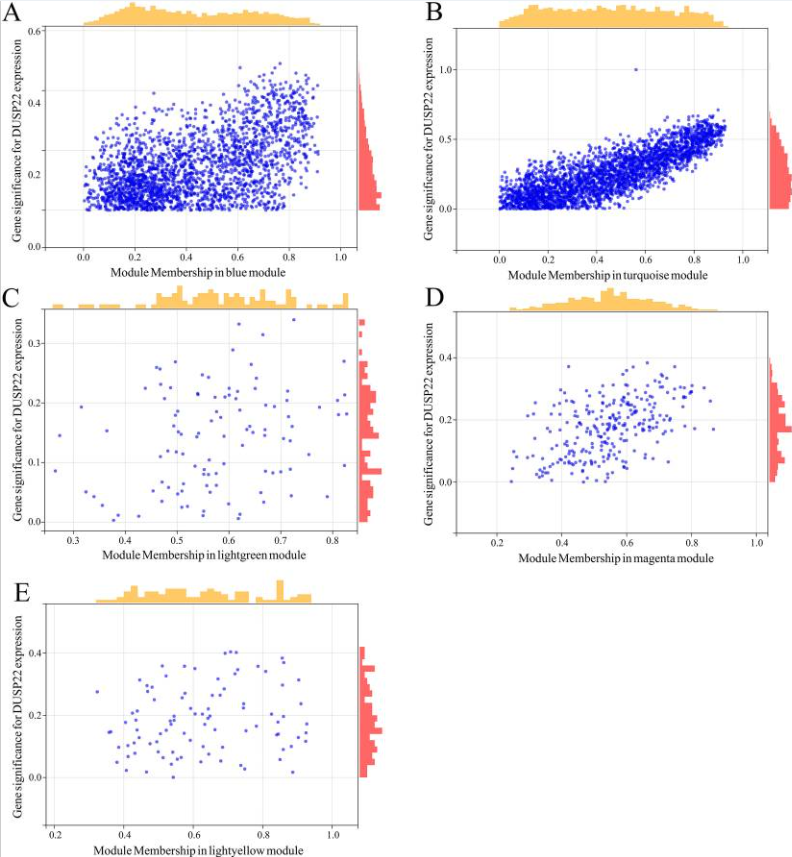
图4 鉴定与USP22共表达的关键基因。
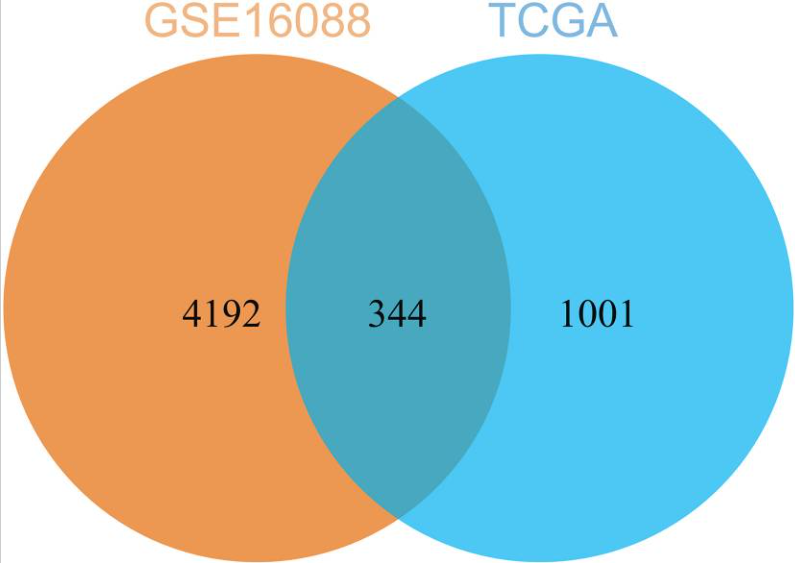
图5通过比较TCGA-OS队列的DEG和数据集中的hub gens GSE16088确定的常见基因。
PPI网络分析与功能富集分析
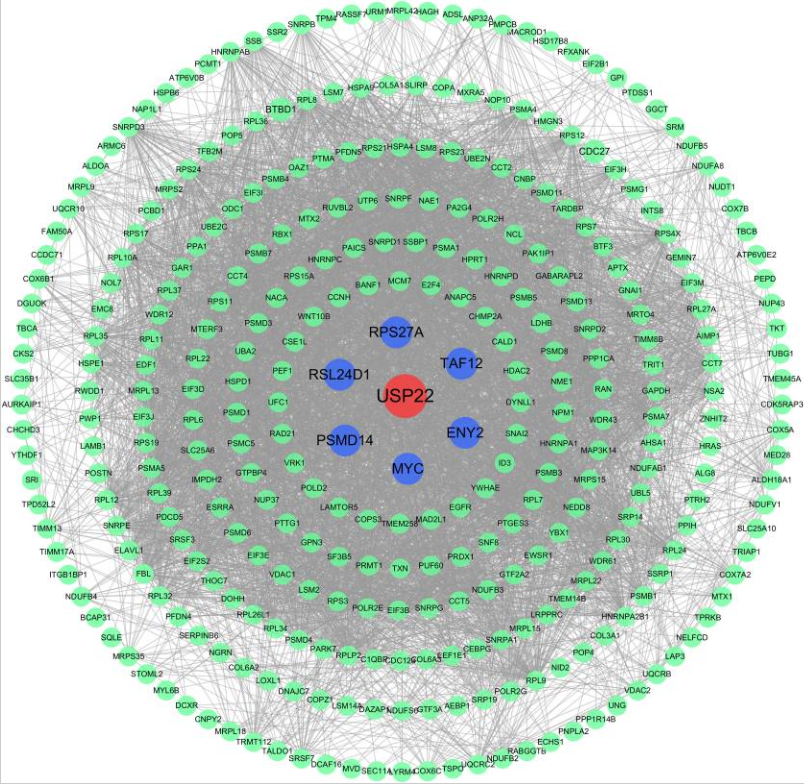
图6 蛋白质-蛋白质相互作用 (PPI) 网络分析。
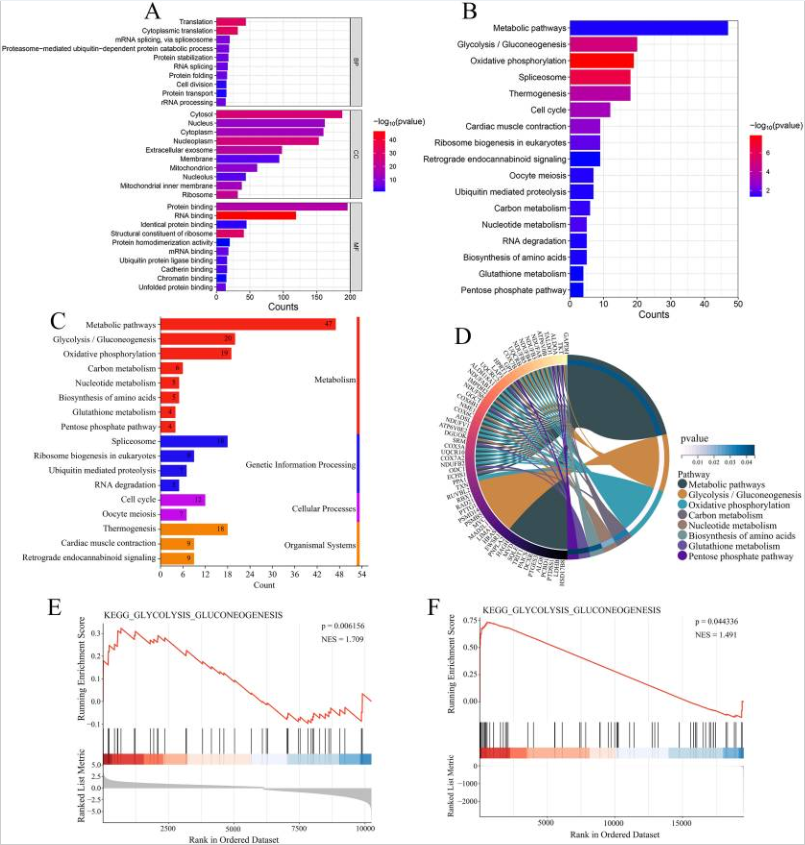
图7 344个共表达基因的功能富集分析。

图8 USP22敲低对骨肉瘤细胞增殖的影响。
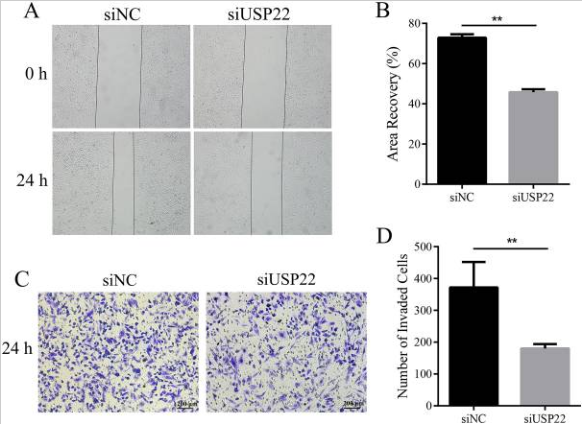
图9 USP22下调对骨肉瘤细胞迁移和侵袭的影响。

图10 USP22下调对骨肉瘤细胞凋亡的影响。
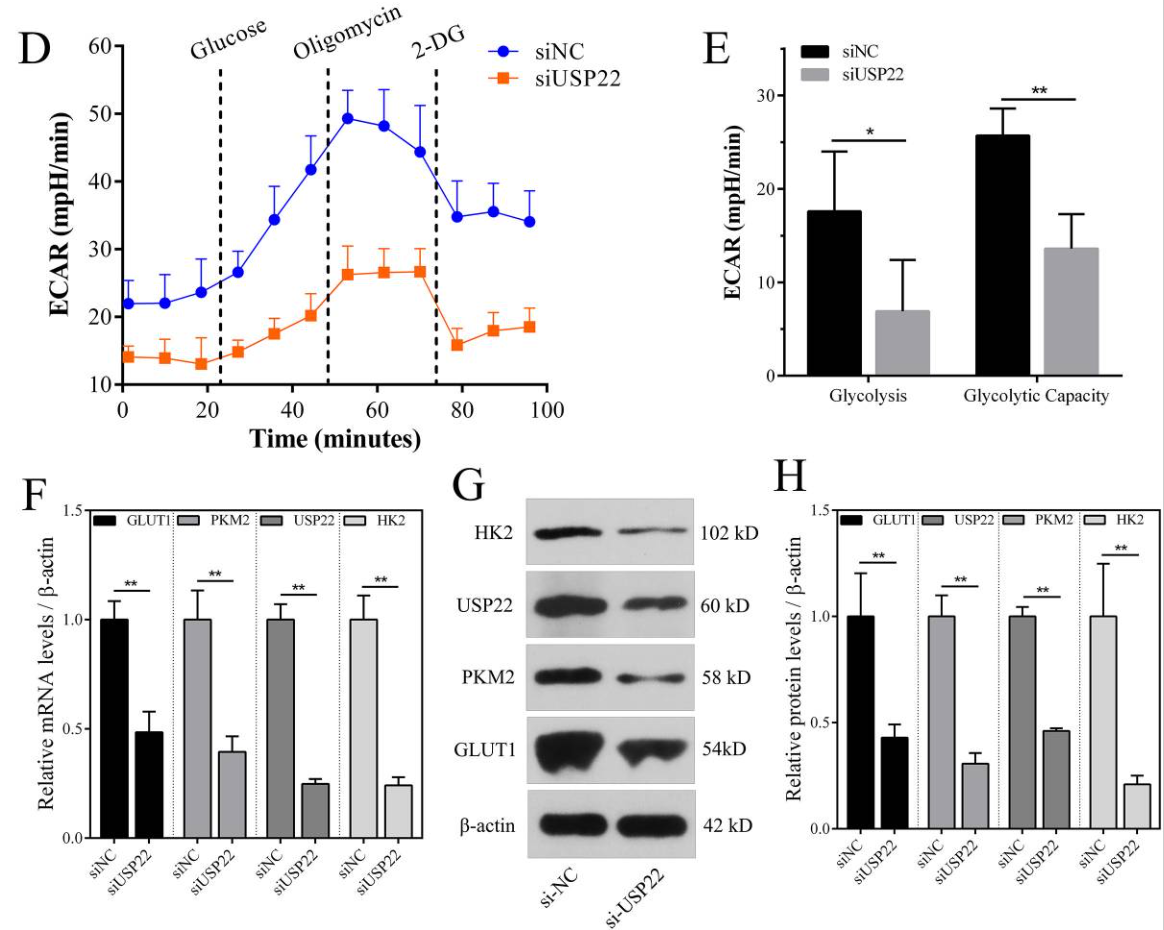
图11 USP22 敲低对骨肉瘤细胞中有氧糖酵解水平的影响。
【结论与讨论】
作者团队研究证明了USP22在骨肉瘤中的致癌作用。根据生物信息学分析,USP22在骨肉瘤中高度表达,并与患者的生存率低有关。USP22促进骨肉瘤的分子机制涉及多种信号通路,包括糖酵解、氧化磷酸化、剪接体、产热和细胞周期。体外实验进一步验证了生物信息学分析结果的准确性。研究表明,USP22的下调可以抑制骨肉瘤细胞的糖酵解和生长。
原文链接:www.ncbi.nlm.nih.gov/pmc/articles/PMC11114114/
大家好!今天小编给大家分享的是一篇于2023年4月同我司合作发表在Frontiers in Immunology杂志(IF: 8.786)的文章《Characterization of the fatty acid metabolism in colorectal cancer to guide clinical therapy》。
背景介绍
肝纤维化(HF)是由各种慢性肝病如病毒性肝炎、酒精性肝炎、胆汁淤积症和药物性肝损伤等引起的过度修复反应所导致的异常细胞外基质沉积。HF已成为全球一个日益严重的公共健康问题,可能导致肝硬化和肝癌等高危严重肝病的发生。然而,目前还没有任何HF治疗药物获临床批准使用。因此,探索HF的发病机制对于发现肝纤维化的靶向治疗药物具有重要意义。HF的直接原因是过量生成和沉积胶原、糖蛋白和蛋白多糖等细胞外基质。HF的发生涉及多个细胞变化。肝星状细胞激活和向肌成纤维细胞转化是HF发展的中心环节。激活的肝星状细胞通过产生大量细胞外基质导致HF的形成。HF过程中常伴随着肝脏炎症损伤。肝脏炎症和肝脏免疫微环境改变被认为是HF发展的关键因素。肝脏含有丰富的免疫细胞,包括先天免疫细胞(库普弗细胞、自然杀伤细胞、自然杀伤T细胞)和获得性免疫细胞(T细胞和B细胞)。越来越多的研究表明,免疫细胞调节HF的进展和消退。在HF过程中,免疫系统通过触发炎症参与组织损伤的修复。肝脏损伤后,肝脏中的免疫细胞被激活并招募到损伤部位,通过分泌肿瘤坏死因子α(TNF-α)、白细胞介素-6(IL-6)、白细胞介素-1β(IL-1β)等促炎细胞因子来激活HSC或损伤的肝细胞。免疫相关基因和信号通路在HF肝组织的免疫细胞浸润中起重要作用。
近年来,随着测序技术的发展,生物信息学已成为广泛用于疾病生物标志物、病理机制和潜在治疗药物识别的良好分析方法。本研究整合生物信息学、转录组学和动物实验,识别了与HF相关的免疫基因和生物通路,有助于我们进一步理解HF的发病机制,为HF的诊断和治疗做出贡献。
全文速览
肝纤维化(HF)的发生和发展伴随着炎症性损害。免疫基因通过调节免疫细胞的浸润,在HF的纤维生成和炎症损害中起着关键作用。然而,对HF的免疫机制的研究还不够充分。因此,本研究旨在确定参与HF纤维化形成和炎症性损害的免疫基因和生物途径,并探索基于免疫靶点的HF治疗方法。研究者从GEO数据库下载HF的表达数据集GSE84044。根据加权基因共表达网络分析(WGCNA)筛选出HF的关键模块基因。将关键模块基因与ImmPort数据库中的免疫相关基因进行比对,得到肝纤维化免疫基因(HFIGs)。此外,研究者对HFIGs进行了基因本体论(GO)和京都基因与基因组百科全书(KEGG)功能富集分析。然后,对HFIGs进行了蛋白质相互作用(PPI)网络,并从PPI网络中确定了hub基因。此外,还进行了免疫浸润分析,以确定hub基因与免疫细胞浸润之间的相关性。为了验证GSE84044表达谱数据分析的可靠性,构建了CCl4诱导的HF大鼠模型,随后对HF大鼠和正常大鼠肝脏组织进行了转录组测序和免疫荧光分析及定量反转录(q-PCR)实验。最后,利用CMAP平台探索基于免疫靶点的HF治疗方法。结果显示,在GSE84044数据的生物信息学分析中,筛选出98个HFIGs,这些基因主要涉及炎症相关的生物途径,如NOD样受体信号通路、NF-kappa B信号通路、Toll样受体信号通路和PI3K-Akt信号通路。从PPI网络中,发现了10个hub基因,包括CXCL8、IL18、CXCL10、CD8A、IL7、PTPRC、CCL5、IL7R、CXCL9和CCL2。免疫浸润分析显示,免疫细胞如中性粒细胞、自然杀伤(NK)细胞、巨噬细胞M1和巨噬细胞M2与肝纤维化过程显著相关,hub基因表达与这些免疫细胞显著相关。值得注意的是,HFIGs富集的大多数生物途径和除CXCL8外的所有hub基因表达都在随后的转录组和qRCR实验中得到了验证。最后,筛选出15个有可能逆转hub基因高表达的小分子化合物作为HF的潜在治疗药物。综上,免疫基因CXCL8、IL18、CXCL10、CD8A、IL7、PTPRC、CCL5、IL7R、CXCL9和CCL2可能在HF的纤维化形成和炎症损害中发挥重要作用。这项研究的成果为研究HF的免疫机制提供了基础,有助于临床实践中对HF的诊断和预防及治疗。
方法解读
1. 从GEO数据库下载基因表达谱数据集GSE84044,包含124例肝纤维化患者的基因表达数据和肝组织炎症分级及纤维化分期等临床信息。
2. 使用WGCNA方法分析GSE84044数据集,设置阈值过滤基因和样本,构建基因共表达网络。计算每个模块与临床特征的相关性,获得与肝纤维化和炎症显著相关的基因模块。计算每个模块的模块成员度和基因显著性,获得与肝纤维化和炎症均显著相关的关键模块基因。
3. 从ImmPort数据库获得免疫相关基因,与WGCNA识别的关键模块基因比较,筛选肝纤维化免疫基因(HFIGs)。
4. 对HFIGs进行GO和KEGG功能富集分析。
5. 通过STRING和Cytoscape构建HFIGs的PPI网络,识别核心基因。
6. 使用CIBERSORT数据库分析GSE84044数据集免疫细胞浸润情况。
7. 建立四氯化碳诱导的大鼠肝纤维化模型,进行组织学、免疫荧光、转录组和qPCR实验验证分析结果。
8. 使用CMAP数据库筛选可能逆转核心基因表达的小分子化合物。
结果分析
1 WGCNA分析和HFIGs鉴定
研究者从GEO数据库下载HF的表达数据集,预处理后,对GSE84044数据进行了WGCNA分析。选择最佳软阈值β为5。然后,基于DynamicTreeCut算法,生成基因模块。如图1C所示,最终产生了21个基因模块。绘制Eigengene树状图和Eigengene邻接图来分析模块Eigengene(ME)的连接性,结果显示模块之间的距离大于0.25。见图1。研究者计算了模块-特征相关系数。结果显示,包括brown4, black, floralwhite, darkturquoise和red在内的5个模块与纤维化和炎症高度相关。因此,研究者选择了与纤维化和炎症显著相关的5个模块进行进一步分析。根据|GS|>0.4和|MM|>0.5以及p<0.05的标准,从5个重要模块中共确定了684个与纤维化和炎症高度相关的关键基因(图2,3)。最后,研究者比较从ImmPort数据库获得的2483个免疫基因,共筛选出98个IRG与纤维化和炎症相关的关键模块基因之间的重叠基因,并确定为肝纤维化免疫基因(HFIGs)(图4)。
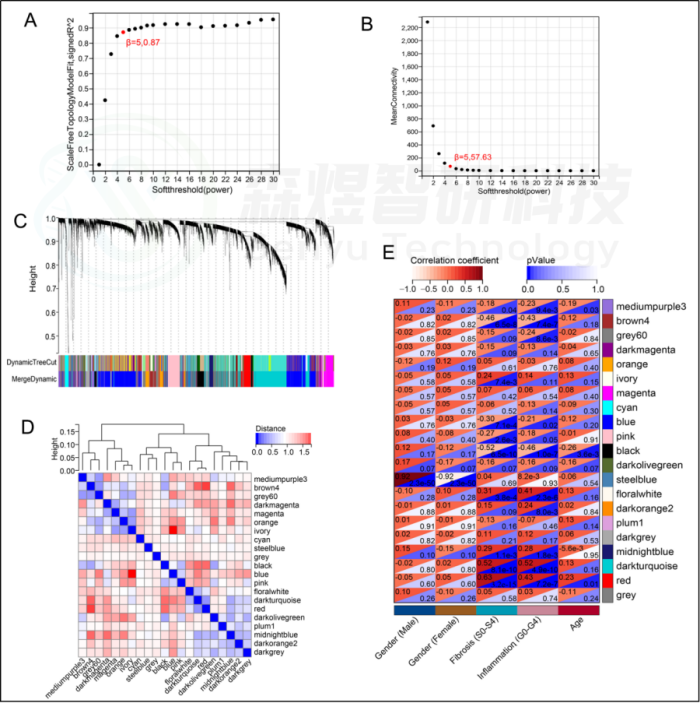
图1.GSE84044数据的WGCNA。(A) 尺度独立性作为软阈值power的函数。(B) 平均连通性与软阈值power的关系。(C) 聚类树状图。每种颜色代表一个特定的共表达模块。聚类树下面的双色行代表原始模块和合并后的模块。(D) Eigengene树状图和模块eigengene邻接(MEs)。(E)MEs和临床性状之间的相关性热图。
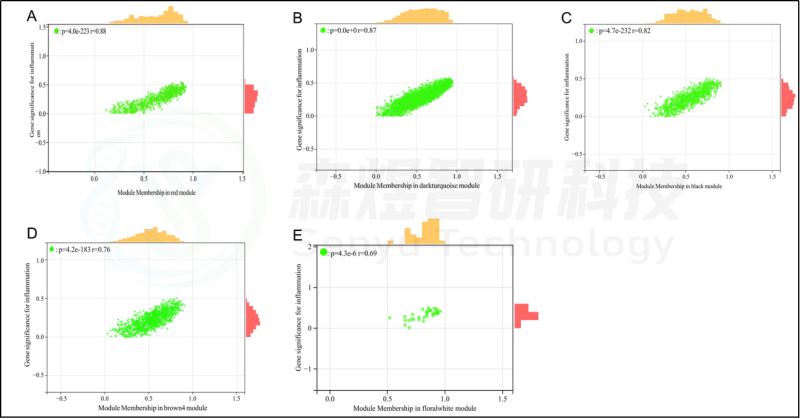
图2.与炎症性状相关的模块的散点图分析。(A-E) 分别是红色、深绿色、黑色、棕色4和花白色的模块的散点图分析。
图3.与纤维化性状相关的模块的散点图分析。(A-E) 红色、深绿色、黑色、棕色4和花白色模块的散点图分析。
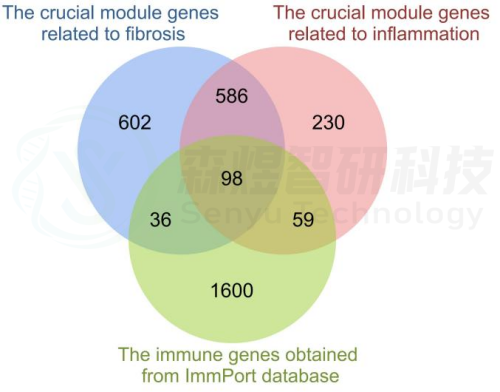
图4.HFIGs鉴定。
2 HFIGs的功能富集分析
为了确定HFIGs的生物学功能,对98个HFIGs进行功能富集分析,包括GO注释和KEGG途径富集分析。GO注释结果显示,98个HFIGs在149个生物过程(BP)、32个细胞成分(CC)和39个分子功能(MF)中显著富集。BP 涉及免疫反应、趋化性、中性粒细胞趋化性、炎症反应、信号转导、趋化因子信号通路、适应性免疫反应、抗原处理和呈递、杀死其他生物体的细胞等。CC包括质膜外侧、细胞外空间、细胞表面、ER至高尔基体运输囊膜、质膜的整体成分等。在MF中,98个HFIGs主要富集在趋化因子活性、肽抗原结合、CCR趋化因子受体结合、MHC II类受体活性、细胞因子活性、CXCR趋化因子受体结合、MHC II类蛋白复合物结合、T细胞受体结合等等。KEGG通路涉及:细胞因子-细胞因子受体相互作用、Th17细胞分化、抗原处理和表达、趋化因子信号通路、TNF信号通路、NF-kappa B信号通路、Toll样受体信号通路等。大多数富集的通路是与免疫有关的通路。见图5。
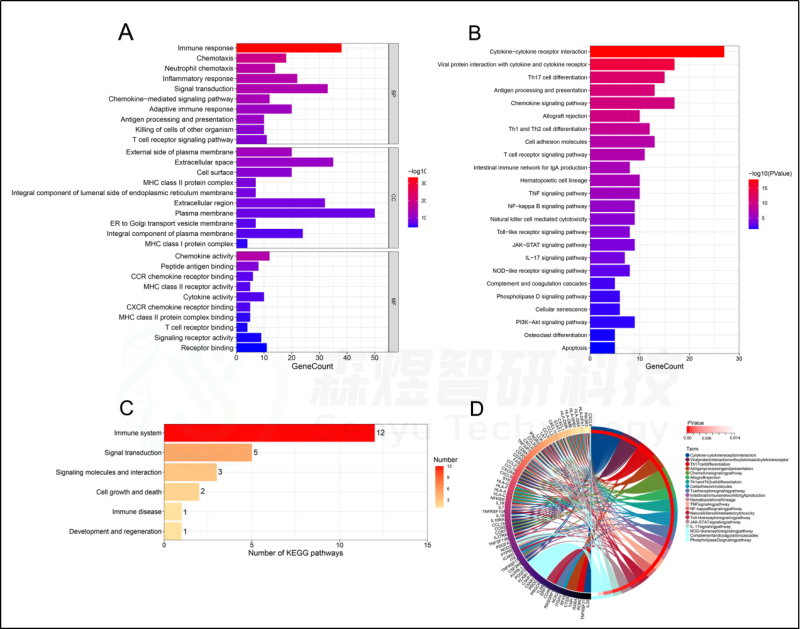
图5.HFIGs的功能富集分析。HFIGs的(A) GO注释。(B) KEGG途径富集分析。(C)KEGG信号通路的分类。(D)KEGG信号通路及其相关的HFIGs的相互作用。
3 PPI网络的构建与hub基因的选择
研究者构建了PPI网络。根据MCC算法,从PPI网络中筛选出排名前10个hub基因,并依次排序如下: CXCL8、IL18、CXCL10、CD8A、IL7、PTPRC、CCL5、IL7R、CXCL9和CCL2。这10个hub基因的表达与纤维化阶段(S0-S4)和炎症等级(G0-G4)呈正相关。所有的hub基因的表达都相互间有显著的相关性。见图6。功能富集分析显示,hub基因富集于多种KEGG途径。与98个HFIGs的KEGG通路富集结果相比,发现了8条共同的信号通路,包括细胞因子-细胞因子受体相互作用、病毒蛋白与细胞因子和细胞因子受体相互作用、趋化因子信号通路、Toll样受体信号通路、NOD样受体信号通路、IL-17信号通路、造血细胞系和TNF信号通路。见图6。
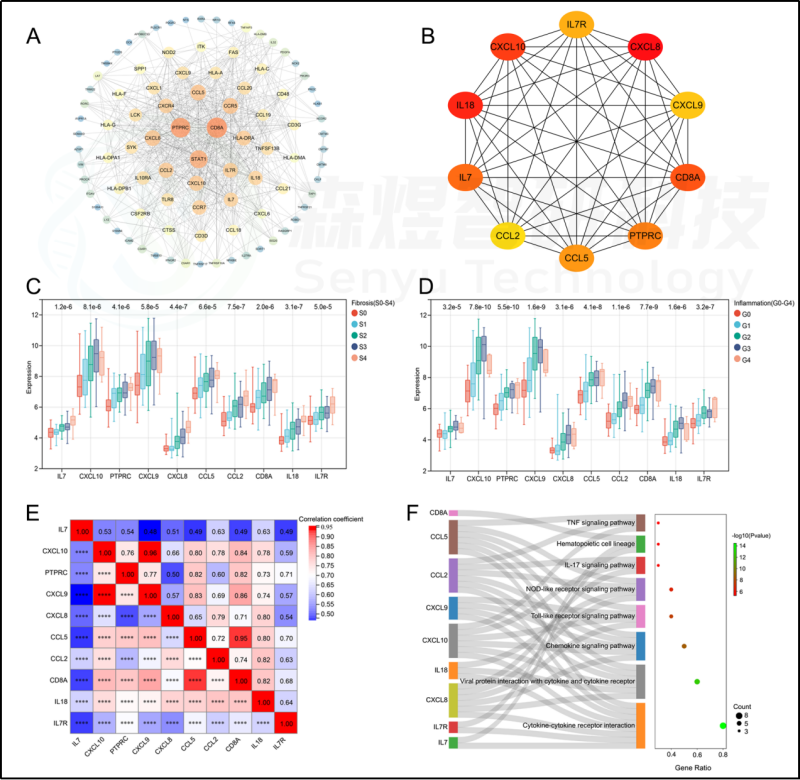
图6.PPI网络构建与hub基因选择。(A) 构建的PPI网络。(B) 10个从PPI网络中鉴定出的hub基因。(C) hub基因在不同纤维化阶段的肝组织中的表达。(D) 不同炎症级别的肝组织中hub基因的表达。(E) hub基因表达的相关性分析。(F) hub基因的功能富集分析。
4 免疫浸润分析
在肝脏组织中发现了21种浸润的免疫细胞类型,除了嗜酸细胞,它们在每个样本中都没有表达。随后,研究者评估了这些免疫细胞群之间的相关性。见图7。研究者还评估了每位患者肝脏组织中21种免疫细胞的浸润水平与纤维化阶段(S0-S4)和炎症等级(G0-G4)之间的相关性。纤维化阶段(S0-S4)与巨噬细胞M1、T细胞gamma delta和B细胞naive的浸润状态呈正相关,与B细胞记忆、巨噬细胞M2、NK细胞静止、中性粒细胞、树突状细胞激活和T细胞调节(Tregs)的浸润状态呈负相关。炎症等级(G0-G4)与T细胞γδ、巨噬细胞M1和B细胞幼稚期呈正相关,而与B细胞记忆期、巨噬细胞M2、NK细胞静止期、肥大细胞静止期、T细胞调节期(Tregs)和中性粒细胞呈负相关。见图8。此外,研究者进一步研究了10个hub基因与21种免疫细胞浸润的相关性。每个hub基因的表达与巨噬细胞M1、T细胞γδ和B细胞的浸润状态呈正相关,而与巨噬细胞M2、B细胞记忆、T细胞调节(Tregs)和NK细胞静止的浸润状态呈负相关。见图9。
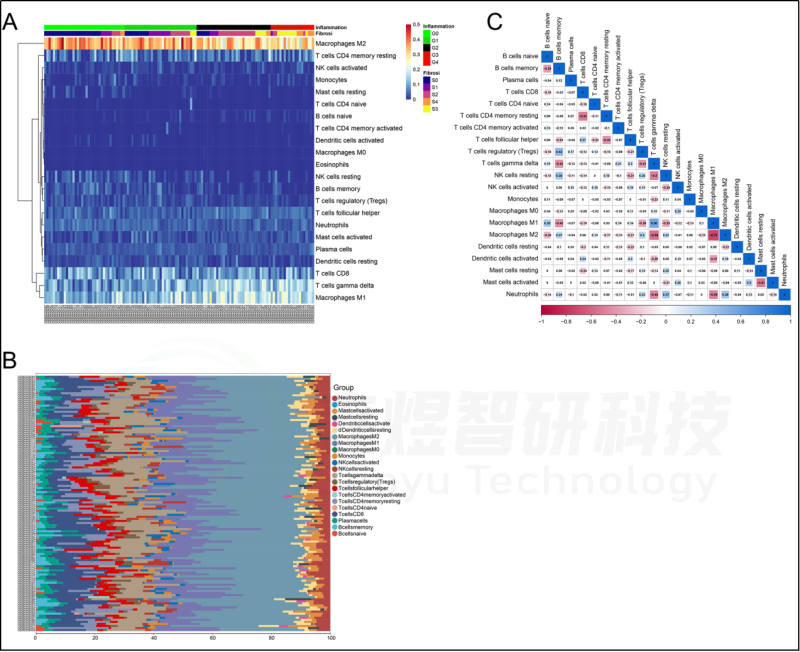
图7.GSE84044数据的免疫浸润分析。(A)129例肝纤维化患者肝组织中免疫细胞浸润的热图。(B)129例肝纤维化患者肝组织中免疫细胞浸润的柱状图。(C)在22种不同的免疫细胞群之间的相关性。
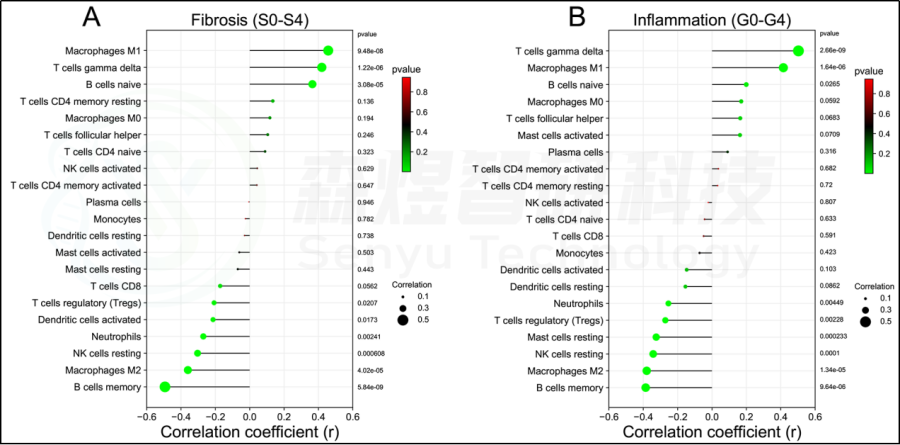
图8.HF患者肝组织纤维化分期与炎症分级与免疫细胞浸润的关系。(A)肝纤维化分期(S0-S4)。(B)肝脏炎症分级(G0-G4)。

图9.肝纤维化患者肝组织中hub基因与免疫浸润的关系。(A) CCL2。(B) CCL5。(C) CD8A。(D) CXCL8。(E) CXCL8。(F) CXCL8。(G) IL7。(H) IL7R。(I) IL18。(J) PTPRC。
5 肝纤维化大鼠模型的建立
为了验证WGCNA分析结果的可靠性,研究者构建了一个HF的大鼠模型。结果显示,模型组大鼠的肝脏组织表现出大量的肝细胞脂肪变性、假小球的形成、肝细胞肿胀和坏死、免疫细胞的浸润和胶原纤维的广泛分布(图10)。ELISA结果显示,与正常对照组相比,CCl4模型组的肝脏HA、LN、IL1β、IL6和TNF-a均显著增加(图11)。
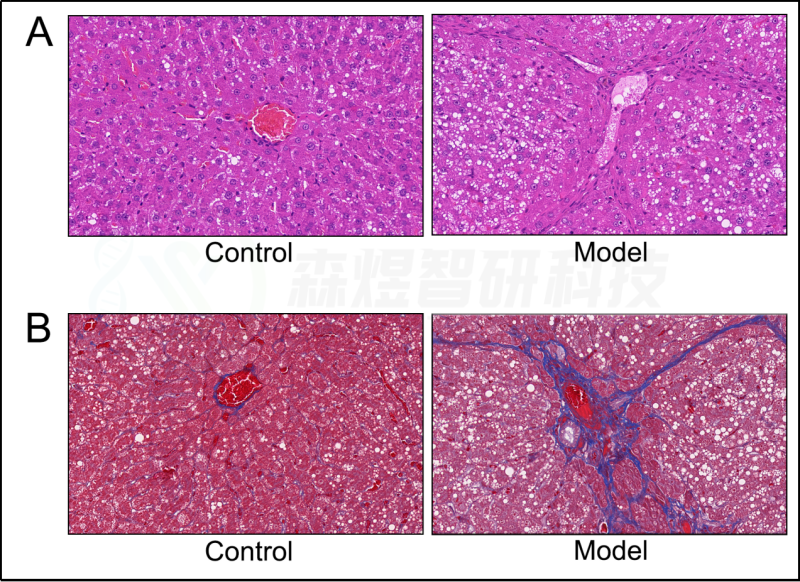
图10.苏木精和伊红(HE)染色和Masson染色。(A) HE染色。(B) Masson染色。
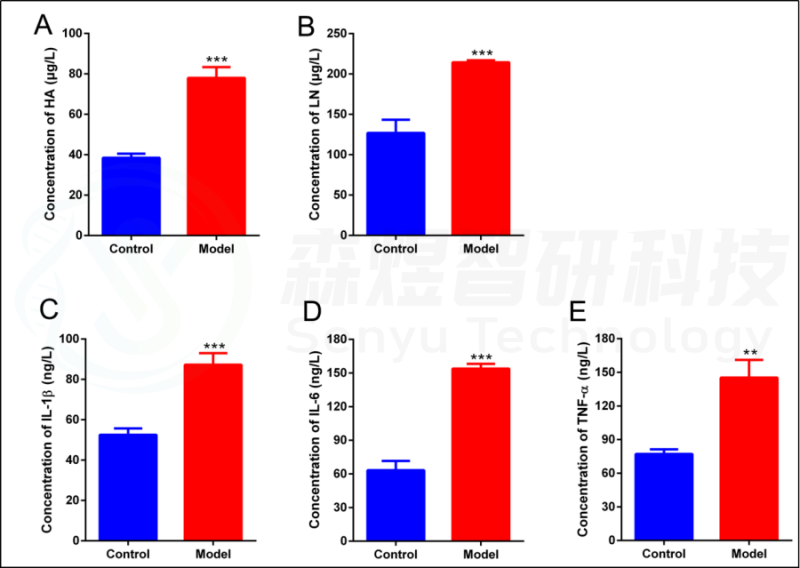
图11.采用ELISA法测定肝组织纤维化及炎症参数。(A) 透明质酸(HA)。(B) 层粘连蛋白(LN)。(C) 白细胞介素1b(IL1b)。(D) 白细胞介素6(IL6)。(E) 肿瘤坏死因子-a(TNF-a)。**p值<0.01;***p-value<0.001,与正常对照组比较。
6 免疫荧光染色
研究者对正常组和模型组的肝组织进行免疫荧光分析。结果显示,Cd86(巨噬细胞M1的标志物)在纤维化大鼠肝脏中的表达水平明显高于正常大鼠肝脏组织,但与正常对照组相比,模型组的Cd10(中性粒细胞的标志物)、Cd11b(NK细胞的标志物)和Cd206(巨噬细胞M2的标志物)表达水平明显下降。免疫荧光染色的结果在一定程度上验证了免疫浸润分析结果的准确性。见图12。
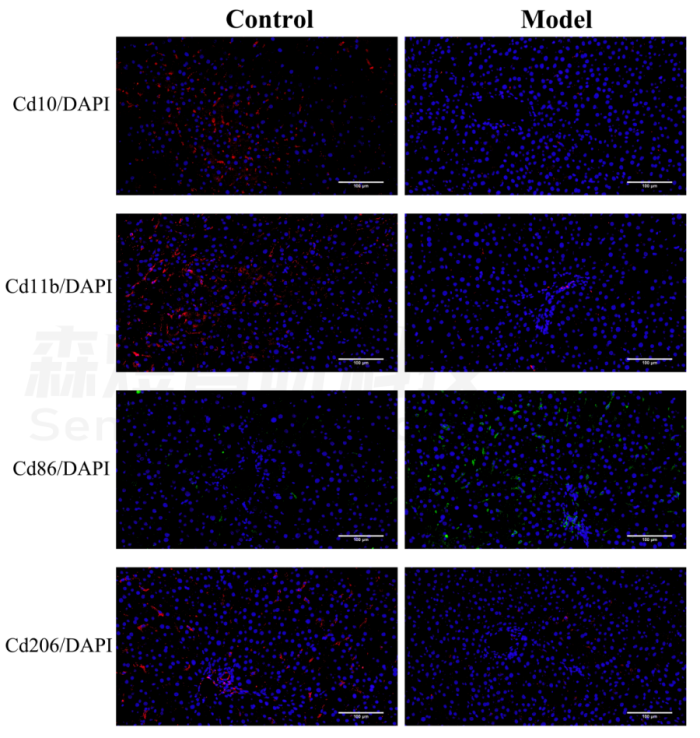
图12.免疫荧光染色检测免疫细胞浸润。
7 RNA-seq分析
研究者利用高通量测序技术对正常大鼠和CCl4诱导的肝纤维化大鼠的肝脏组织进行了检测,然后对差异基因进行筛选。筛选出1039个被注释的差异基因。主要富集在14条KEGG通路中,这些通路在之前的研究结果中被确定为HFIGs相关的KEGG信号通路,包括破骨细胞分化、趋化因子信号通路、自然杀伤细胞介导的细胞毒性、补体和凝血级联、Toll样受体信号通路、PI3K-Akt信号通路、NOD样受体信号通路、凋亡、NF-κB信号通路等。此外,研究者利用GSEA研究了在模型组和正常对照组中显著差异表达的信号通路。与前述结果中富含HFIGs的24条通路相比,研究者发现了19条共同的KEGG通路。如表1所示,与正常对照组相比,所有的共同途径在模型组都有显著的高表达。
研究者从RNA-seq数据中筛选出9个hub基因,包括Il18、Cxcl10、Cd8a、IL7、Ptprc、Ccl5、Il7r、Cxcl9和Ccl2(Cxcl8在大鼠物种中没有表达,所以不检测)。与正常肝脏组织相比,CCl4诱导的肝纤维化大鼠肝脏组织中的9个hub基因显著上调。Person相关分析显示所有9个hub基因之间有良好的相关性。总的来说,研究者的转录组分析结果进一步证实了这些免疫相关信号通路和hub基因在HF进展中的关键作用。见图13。
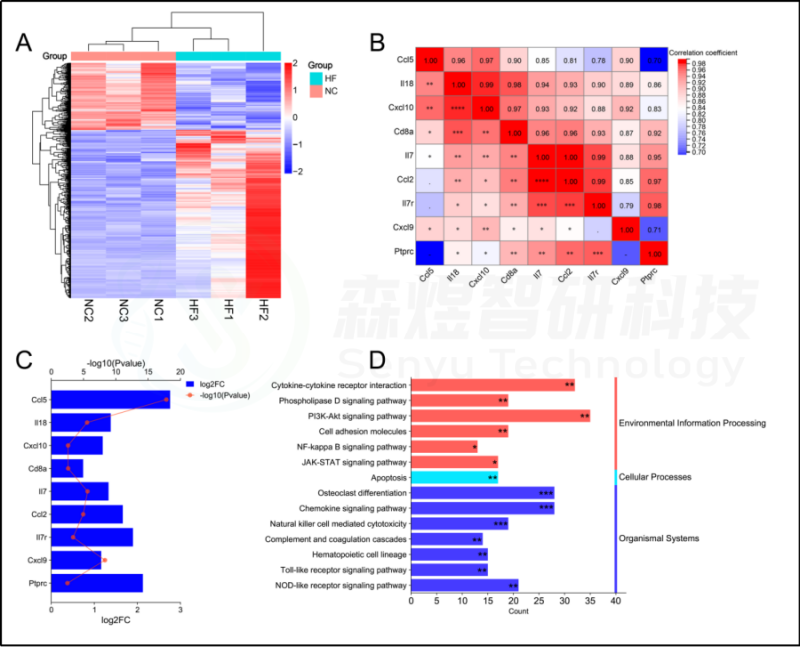
图13.RNA-seq数据的生物信息学分析。(A) RNA-seq数据中差异表达基因(DEGs)的热图。(B) RNA-seq数据中hub基因表达的相关性分析。(C) RNA-seq数据中正常组和CCl4模型组之间hub基因表达的变化。(D) 通过DAVID数据库富集的HFIGs相关和DEGs相关的共同信号通路。*p-value<0.05;**p-value<0.01;***p-value<0.001,表示功能富集结果的意义。
表1.GSEA富集的DEGs与DAVID数据库富集的HFIGs之间的共同信号通路。
ID | Description | Enrichment Score | NES | pvalue | qvalue |
ko04621 | NOD-like receptor signaling pathway | 0.52 | 1.72 | 1.07E-03 | 7.94E-03 |
ko04062 | Chemokine signaling pathway | 0.58 | 1.87 | 1.09E-03 | 7.94E-03 |
ko04060 | Cytokine-cytokine receptor interaction | 0.57 | 1.83 | 1.10E-03 | 7.94E-03 |
ko04514 | Cell adhesion molecules | 0.58 | 1.88 | 1.10E-03 | 7.94E-03 |
ko04380 | Osteoclast differentiation | 0.65 | 2.08 | 1.12E-03 | 7.94E-03 |
ko04064 | NF-kappa B signaling pathway | 0.60 | 1.87 | 1.14E-03 | 7.94E-03 |
ko04612 | Antigen processing and presentation | 0.61 | 1.88 | 1.14E-03 | 7.94E-03 |
ko04650 | Natural killer cell mediated cytotoxicity | 0.75 | 2.32 | 1.15E-03 | 7.94E-03 |
ko04658 | Th1 and Th2 cell differentiation | 0.63 | 1.91 | 1.19E-03 | 7.94E-03 |
ko04640 | Hematopoietic cell lineage | 0.73 | 2.22 | 1.19E-03 | 7.94E-03 |
ko05330 | Allograft rejection | 0.76 | 2.16 | 1.25E-03 | 7.94E-03 |
ko04061 | Viral protein interaction with cytokine and cytokine receptor | 0.69 | 1.92 | 1.27E-03 | 7.94E-03 |
ko04672 | Intestinal immune network for IgA production | 0.81 | 2.15 | 1.31E-03 | 7.94E-03 |
ko04072 | Phospholipase D signaling pathway | 0.55 | 1.76 | 2.20E-03 | 1.19E-02 |
ko04620 | Toll-like receptor signaling pathway | 0.52 | 1.62 | 4.55E-03 | 1.89E-02 |
ko04659 | Th17 cell differentiation | 0.50 | 1.57 | 6.77E-03 | 2.50E-02 |
ko04660 | T cell receptor signaling pathway | 0.48 | 1.49 | 1.26E-02 | 4.17E-02 |
ko04151 | PI3K-Akt signaling pathway | 0.39 | 1.33 | 1.64E-02 | 5.02E-02 |
ko04610 | Complement and coagulation cascades | 0.44 | 1.38 | 3.64E-02 | 9.01E-02 |
8 通过q-PCR验证hub基因的表达
由于Cxcl8在大鼠体内不表达,因此,研究者通过q-PCR实验检测了包括Il18、Cxcl10、Cd8a、IL7、Ptprc、Ccl5、Il7r、Cxcl9和Ccl2在内的9个hub基因的mRNA表达水平,以验证正常组和模型组肝组织中确定的hub基因的表达。结果显示,与正常对照组相比,模型组中hub基因的mRNA表达显著上调,与之前的WGCNA分析和RNA测序结果一致。
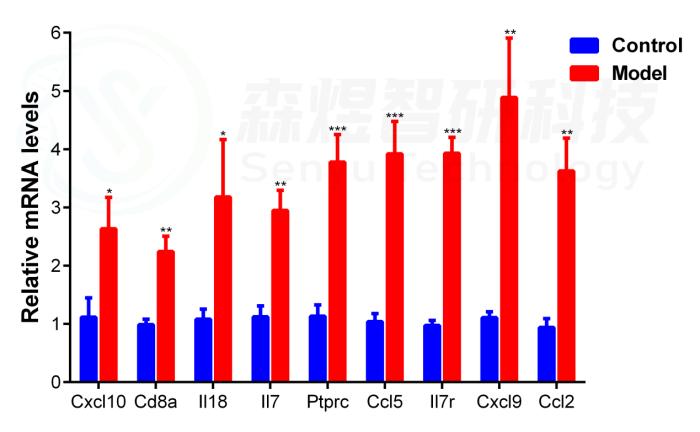
图14.通过q-PCR检测正常组和四氯化碳模型组中9个hub基因的mRNA表达量。*p-value< 0.05;**p-value<0.01;***p-value<0.001,与正常对照组比较。
9 候选小分子的鉴定
研究者通过CMAP筛选逆转HF的潜在药物。筛选出连通性分数为负的前15个小分子化合物,包括elotristat, lomitapide, malotilate, tranylcypromine, TG-101348, cordycepin, tafamidis-meglumine, fostamatinib, delcorine, phenylbutazone, endo-IWR-1, scopolamine, anidulafungin, simvastatin 和 terconazole(表2)。这些小分子化合物有可能通过抑制hub基因的上调而发挥抗肝纤维化的作用。
表2.小分子鉴定结果。
Rank | CMAP name | Connectivity score | -log (p value) |
1 | telotristat | -0.912 | 15.6536 |
2 | lomitapide | -0.883 | 15.6536 |
3 | malotilate | -0.8735 | 15.6536 |
4 | tranylcypromine | -0.8728 | 15.6536 |
5 | TG-101348 | -0.8707 | 15.6536 |
6 | cordycepin | -0.8564 | 15.6536 |
7 | tafamidis-meglumine | -0.8454 | 12.2035 |
8 | fostamatinib | -0.8445 | 11.9793 |
9 | delcorine | -0.8442 | 2.9231 |
10 | phenylbutazone | -0.8437 | 2.8391 |
11 | endo-IWR-1 | -0.8432 | 2.7737 |
12 | scopolamine | -0.8392 | 2.4983 |
13 | anidulafungin | -0.8386 | 1.9741 |
14 | simvastatin | -0.8371 | 1.9178 |
15 | terconazole | -0.835 | 1.8569 |
结论
本研究表明,免疫机制在HF的发生和发展中起着关键作用,并探索了基于免疫靶点的HF治疗方法。研究者筛选了98个与HF相关的免疫基因,并从中确定了10个hub基因,包括CXCL8、IL18、CXCL10、CD8A、IL7、PTPRC、CCL5、IL7R、CXCL9和CCL2。研究者在HF大鼠的肝脏组织中检测了这9个hub基因的表达,结果表明这些hub基因可能对HF的纤维化形成和炎症性损伤有贡献。研究者确定了几个与HF相关的免疫信号通路,如NOD样受体信号通路、NF-kappa B信号通路、Tolllike受体信号通路、PI3K-Akt信号通路、T细胞受体信号通路和磷脂酶D信号通路。免疫浸润分析和免疫荧光分析表明,免疫细胞如中性粒细胞、NK细胞、巨噬细胞M1和巨噬细胞M2的浸润状况与HF显著相关。综合来看,本研究揭示了HF的潜在免疫机制,并研究了逆转HF的免疫治疗药物,该疾病的具体分子机制和小分子药物的药理机制还需要进一步探索。
原名:Revealing immune infiltrate characteristics and potential immune-related genes in hepatic fibrosis: based on bioinformatics,transcriptomics and q-PCR experiments
译名:基于生物信息学、转录组学和q-PCR实验的结果揭示肝纤维化中的免疫浸润特征和潜在的免疫相关基因
期刊:Frontiers in Immunology
IF:8.786
发表时间:2023年4月
DOI号:10.3389/fimmu.2023.1133543
大家好!今天小编跟大家分享的是一篇于2023年1月同我司合作发表在Biochemical Genetics杂志(IF: 2.4)的文章案例《Identification of lncRNA-miRNA-mRNA Regulatory Network and Therapeutic Agents for Skin Aging by Bioinformatics Analysis》。
皮肤衰老是人体老化最直观的表现,不仅可能导致美观和心理问题,甚至转化成疾病。本研究旨在研究皮肤衰老的病理和分子机制,并确定逆转皮肤衰老的治疗药物。对皮肤衰老基因表达数据集(GSE55118和GSE72264)进行生物信息学分析。首先,通过WGCNA网络分析识别出与皮肤衰老密切相关的169个关键mRNAs、27个关键miRNAs和50个关键lncRNAs。然后,通过Metascape数据库执行的功能富集分析显示,皮肤衰老涉及多种生物功能,如刺激检测、对类固醇激素的响应、水通道活性、肌肉收缩的调节。接下来,通过PPI相互作用分析筛选出10个中心基因,包括AQP4,TRPM8,TBR1,NTSR2,MPPED1,BARHL2,PAX9,CPN1,CES3,和CHGB。接下来,构建了“lncRNA-miRNA-mRNA”网络和“lncRNA-miRNA-中心mRNA”网络,以探索皮肤衰老的竞争性内源性RNA机制。最后,通过CMAP平台筛选出10种显著的可能缓解皮肤衰老的小分子,包括维生素A酸、吡非胺、司拉克丁、恩替诺特、溴他西尼、丁香酸、BRD-K9647565、依美斯汀、阿巴卡韦和鱼腾酮,并通过分子对接实验验证了它们的可靠性。本研究为揭示皮肤衰老的分子机制提供了基础,并确定了可能的缓解皮肤衰老的候选药物。
作为人体最大的器官,皮肤会随着生物学年龄的增长而表现出老化。随着人类寿命的延长,老龄化社会的到来以及公众对美的认识,探索皮肤衰老的原因以及如何延缓其老化过程已经成为化妆医学领域的一个热点。此外,随着人们年龄的增长,人类皮肤的形态结构和生理功能会发生退行性变化。皮肤衰老是许多疾病的风险因素。老年人的皮肤容易干燥、发痒、色素沉着、皱纹以及良性和恶性肿瘤。因此,保持我们的皮肤健康有助于促进健康老龄化。
皮肤衰老是一个复杂且持续的生物学过程,其特点包括表皮变薄、弹性组织退化、黑色素细胞数量减少和屏障功能受损,表现为皱纹、弹性减退、干燥和色素沉着障碍。许多内在和外在因素都会导致皮肤衰老。外源性老化主要是由电离和非电离辐射、空气污染、自然有害气体(例如臭氧和高氧浓度)、吸烟、病原细菌入侵、病毒以及其他因素引起的。其中,长期反复暴露于阳光中的紫外线(UV)是环境中影响皮肤衰老的最重要因素。此外,内源性老化,也称为自然老化,是由内在因素引起的衰老,包括衰老、生理老化、皮肤合成胶原蛋白和弹性蛋白的能力下降,以及细胞质和细胞间基质的活动性下降。
皮肤衰老的机制复杂,许多理论已被提出来解释老化的机制,如老化基因理论、自由基理论和线粒体理论。近年来,随着测序技术的发展,通过比较年轻人和老年人之间的皮肤差异,已经鉴定出许多老化基因,这为研究皮肤衰老生物标志物和分子机制提供了基础。作为皮肤衰老的一个重要生物标记,已发现水通道蛋白(AQPs)的低表达是皮肤衰老的风险因素,并且 AQPs 通过调节皮肤表皮细胞的增殖和分化可以延缓皮肤的过早衰老。
竞争内源性RNA(ceRNA)是竞争与同一miRNA结合的转录本,随后调控相关靶基因的表达,在生理和病理状态下的许多细胞过程中发挥重要作用。根据ceRNA理论,lncRNA可以作为miRNA海绵调控mRNA表达,调节下游分子过程(lncRNA表达分析冠心病患者心外膜脂肪组织和相关的ceRNA网络分析)。已有研究证明,ceRNA机制是皮肤衰老的重要分子机制。例如,研究者发现“lncRNA-miRNAmRNA”信号可能通过调节TNF信号通路和甲状腺激素信号通路来影响皮肤衰老。LncRNA RP11-670E13.6可作为海绵结合microRNA-663a响应元件,进一步调节Cdk4和Cdk6的表达,诱导皮肤损伤延迟细胞衰老。然而,关于ceRNA参与皮肤衰老的机制的研究仍然很少。因此,深入研究ceRNAs的调控机制,有助于揭示皮肤衰老的调控机制。
近年来,药学科学家在现有药物知识的基础上大力开发新的抗衰老药物。然而,由于成本高、耗时工作,大多数时间无法进行体外或体内研究。相反,由于最近在基于细胞的表型筛选和大量基因组数据分析方面的技术进步,基于生物信息学分析的虚拟筛选为新药的开发提供了便利。连接图(Cmap)包含了经生物活性小分子处理的人类细胞的转录组数据,是一个可靠的生物信息学程序,用于预测各种疾病的潜在药物。
本文利用微阵列数据研究了与皮肤衰老密切相关的基因,然后探索了这些基因的生物学功能,并构建了ceRNA网络。研究结果将在一定程度上揭示皮肤衰老的分子机制。此外,Cmap作为一种有效的药物基因组药物发现工具,被用于筛选潜在的缓解皮肤衰老的候选药物。
1. 从GEO数据库下载两个GSE片段(GSE55118和GSE72264)。GSE55118数据是lncRNA和mRNA数据集,包含5名年轻健康志愿者(20-30岁)和5名中年健康志愿者(40-50岁)以及5名老年健康志愿者(60-70岁)的皮肤标本。GSE72264是一个miRNA数据集,包含来自6名年轻健康志愿者(10岁以下)和6名老年健康志愿者(60岁以上)的皮肤标本。
2. 使用WGCNA方法分别对GSE55118和GSE72264数据集进行分析,设置合适软阈值β过滤基因和样本,构建基因共表达网络。计算每个模块与临床特征的相关性,获得与皮肤衰老高度相关的基因模块并鉴定出关键基因。
3. 将GSE55118中鉴定的关键基因用于GO注释、KEGG通路、维基通路和贺曼基因集富集分析。
4. 使用STRING和Cytoscape构建GSE55118数据集中关键mRNA的PPI网络,并基于MCC算法识别10个核心基因。
5. 使用miRTarbase工具和miRNet数据库预测加权关键lncRNA和基因共表达网络分析筛选的mRNA之间的相互作用,并构建ceRNA网络。数据可视化采用Cytoscape软件。
6. 使用CMap数据库筛选可能逆转与皮肤衰老有关的核心基因表达的小分子化合物。
7. 使用Discovery Studio 2019 software 验证小分子药物的预测结果的可靠性。
1 WGCNA识别关键的mRNA和LncRNA
作者首先将GSE55118的数据经过标准化(图1A)和差异分析,并通过WGCNA构建共表达模块。选择最佳软阈值β为9(图1B)。这些具有相似表达模式的基因被放入一个模块中。经过层次聚类和模块合并,确定10个模块(图1C)。分析了这10个基因共表达模块之间的相互作用关系,结果表明,不同模块之间的关系没有任何明显的差异,表明模块和每个模块中的基因是独立的(图1D)。然后,对模块特征基因(MEs)的连通性进行分析,结果显示每个模块具有较高的特殊性(不相似度<0.2)(图1E)。关于模块-性状相关系数发现,除灰色模块外,所有模块都与皮肤衰老高度相关(p value<0.05)(图1F)。
以|GS|>0.85和|MM|>0.85和p value<0.05作为揭示关键基因的标准,共鉴定出219个与皮肤衰老高度相关的关键基因,包括169个关键mRNA和50个关键lncRNA。在这些关键基因中,包含23个lncRNA和91个mRNA的114个基因的表达与皮肤衰老呈正相关,而包含27个lncRNA和78个mRNA的105个基因的表达与皮肤衰老呈负相关(图2)。
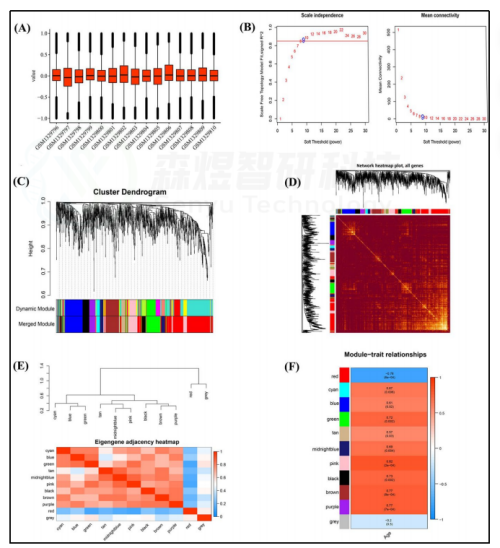
图1. GSE55118的WGCNA (A) GSE55118数据的标准化。(B)软阈值β和无标度网络建设。(C) 聚类树状图。(D) 拓扑重叠测量(TOM)矩阵的热图。(E) 基因共表达网络中邻接点的热图。(F) 模块特征基因与临床特征(年龄)相关性的热图。

图2. (A) GSE55118数据集169个关键mRNA的表达热图。(B) GSE55118数据集中50个关键lncRNA的热图。红点表示高表达RNA,绿点表示低表达RNA
2 功能富集分析
使用Metascape数据库对GSE55118中识别的219个关键基因(169个关键mRNA和50个关键lncRNA)进行了功能富集分析。结果显示,这些关键基因富集于191个生物学过程、33个细胞组分、33个分子功能、7个基因集 Hallmark、8个KEGG通路和9个Wiki通路。关键基因主要参与对刺激的检测、对类固醇激素的响应、水通道活性、肌肉收缩的调节、受体配体活性、解剖结构大小的调控、细胞顶端部分、过渡金属离子运输、核受体激素活性、心血管疾病发病假说通路、穿膜钙离子运输、对外部刺激的检测、眼感光受体细胞发育、淋巴细胞激活的调节、前脑发育、防御反应的调节、蛋白多糖结合、ERAD通路、肌肉收缩的正调控、膜固定组分(图3)。
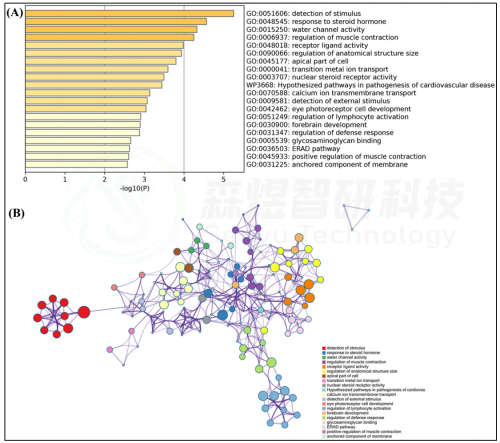
图3. GSE55118中识别出的关键基因的功能富集分析 (A) 富集度排名前20的条形图 (B) 富集项的网络
3 PPI网络的构建与hub基因的选择
作者将WGCNA网络中识别出的169个关键mRNA用于进行PPI网络分析(图4A)。根据MCC算法,从CytoHubba插件的PPI网络中筛选出十个顶级核心基因,顺序排列如下:AQP4、TRPM8、TBR1、NTSR2、MPPED1、BARHL2、PAX9、CPN1、CES3和CHGB(图4B)。功能富集分析显示,10个hub基因富集于10个生物学过程,包括对皮质醇的响应、对皮质类固醇的响应、对类固醇激素的响应、对激素的响应、细胞化学稳态、细胞稳态、细胞大小的调节、细胞组分大小的调节、解剖结构大小的调节和细胞命运决定,以及1个基因集Hallmark,即小分子的运输(图4C)。
在10个核心mRNA中,9个核心mRNA的表达与年龄呈负相关,包括AQP4、TRPM8、TBR1、NTSR2、MPPED1、BARHL2、PAX9、CES3和CHGB。核心基因CPN1在中年组或老年组中均显著上调(图5)。
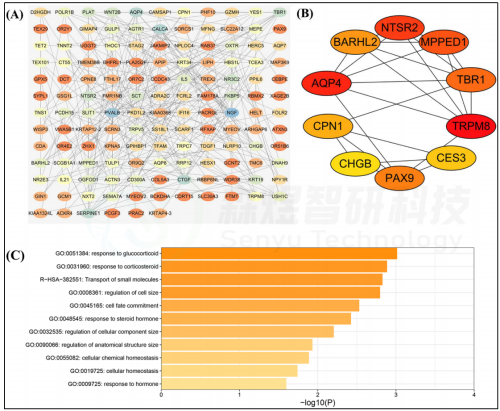
图4. 蛋白互作网络构建和核心基因筛选 (A) 蛋白质互作网络。颜色越深的节点在网络中的度数分值越高。(B) 从蛋白质互作网络中筛选出的10个核心基因。节点根据MCC评分排名,颜色越深红表示MCC评分越高。(C) 10个核心基因的功能富集分析
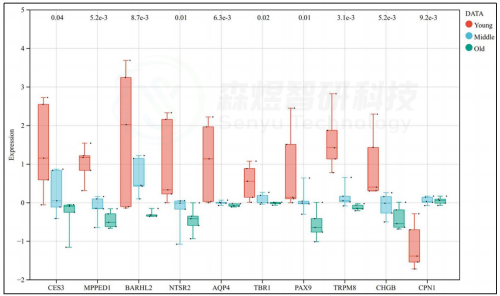
图5. Hub基因在GSE55118数据集中的表达情况
4 WGCNA识别关键的miRNAs
作者对GSE72264基因表达矩阵中的所有1350个基因都用于基因WGCNA分析以便筛选出关键miRNAs(图6A)。最佳软阈值β设置为9(图6B)。按动态剪枝树进行模块赋值后,构建了7个模块,包括蓝色、棕色、绿色、灰色、红色、青绿色、黄色模块,分别包含86、39、37、66、36、1048和37个基因(图6C)。模块及每个模块中的基因是独立的(图6D)。基于ME聚类树和热图结果,表明模块之间的连接性不强(图6E)。在7个模块中,蓝色模块与皮肤衰老显著相关(r=0.94和p值=4e-06)(图6F)。
从蓝色模块中,GS>0.85和|MM|>0.85且p值<0.05的27个miRNA被定义为皮肤衰老发展中的关键miRNA。作者进一步分析了这些关键miRNA在年轻组和老年组中的差异表达。结果显示,与年轻组相比,老年组中6个关键miRNA下调,21个关键miRNA上调(图7)。
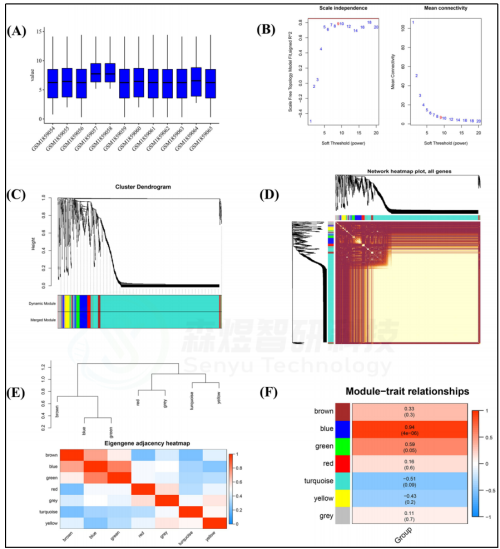
图6. GSE72264的WGCNA分析 (A)GSE72264数据的标准化 (B)软阈值功率筛选和无尺度网络构建 (C)聚类树形图 (D)网络拓扑重叠度量(TOM)矩阵的热图 (E)基因共表达网络中邻接矩阵的热图 (F) 模块特征基因与临床特征(年龄)之间相关性的热图
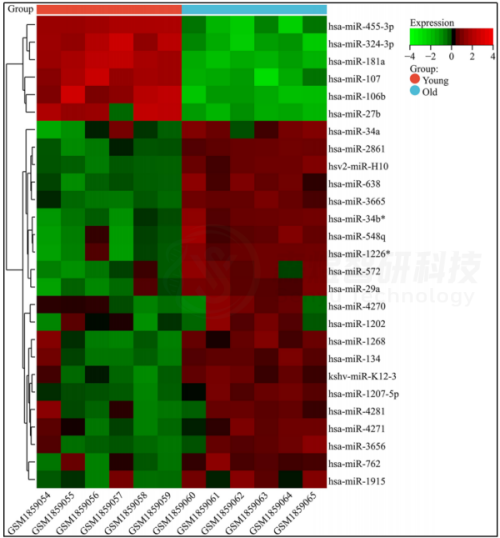
图7. GSE55118数据集中27个关键miRNA的表达热图。红色表示高表达,绿色表示低表达
5 ceRNA网络的建设
为了预测目标基因和lncRNA,作者将27个关键miRNA上传到miRTarBase和TargetScan数据库。然后,与GSE55118中识别的169个关键mRNA和50个关键lncRNA进行比较,识别出63个共同mRNA和6个共同lncRNA,随后识别出与共同mRNA和lncRNA作用的9个关键miRNA。共识别出6个关键lncRNA、9个关键miRNA和63个关键mRNA之间的相互作用,并使用Cytoscape软件构建“lncRNA-miRNA-mRNA”网络。“lncRNA-miRNA-mRNA”网络包含136对miRNA-mRNA相互作用和19对miRNA-lncRNA相互作用(图8A)。
由于中心基因在PPI网络中起着关键作用,中心基因的ceRNA机制可能在皮肤衰老的进展和逆转中发挥重要作用。因此,从“lncRNA-miRNA-mRNA”网络中识别出中心mRNA及其相关miRNA和lncRNA之间的相互作用,构建“lncRNA miRNA中心mRNA”网络。“lncRNA miRNA中心mRNA”网络由9个lncRNA、6个miRNA和4个mRNA组成,包含22对交互关系,包括8对miRNA-mRNA相互作用和14对miRNA-lncRNA相互作用(图8B)。

图8. ceRNA网络构建 (A) “lncRNA-miRNA-mRNA”网络。红色四边形代表lncRNA,蓝色菱形代表miRNA,绿色椭圆代表mRNA (B) “lncRNA-miRNA-中心mRNA”网络的桑基图。
6 药物筛选与分子对接
为了筛选可能逆转皮肤衰老的潜在药物,作者将GSE55118中识别的169个关键mRNA上传到CMAP。前10个最显著的潜在小分子药物是维生素A酸、吡非胺、司拉克丁、恩替诺特、溴他西尼、丁香酸、BRD-K9647565、依美斯汀、阿巴卡韦和鱼腾酮(表1)。
为了探索上述通过筛选获得的小分子是否能直接靶向与皮肤衰老相关的基因,作者进一步选择了hub基因AQP4用于分子对接分析。Discovery Studio 2019软件计算的LibDock Score和AutoDock Vina软件计算的结合能量显示在表2中。所有小分子与AQP4的相互作用的LibDock Score≥70,所有结合能量≤-5.0 kcal/mol。结果表明这些分子与AQP4具有良好的靶向潜力。结合能量最低的5个对接结果显示在图9中。分子对接的结果在一定程度上验证了药物预测的准确性。
表1 模块基因的小分子鉴定(前10位)

表2 10个小分子与AQP4之间的分子对接
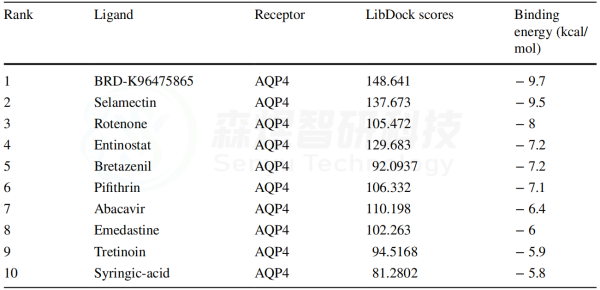

图9. 与AQP4对接的5个最相关小分子的分子对接 (A) BRD-K96475865 (B) 司拉克丁 (C) 鱼腾酮 (D) 恩替诺特 (E)溴他西尼
结论
本研究通过WGCNA网络分析识别了与皮肤衰老密切相关的169个关键mRNA、27个关键miRNA和50个关键lncRNA。这些关键基因可能作为皮肤衰老的生物标志物,并为揭示皮肤衰老的分子机制提供依据。研究者确定了一些与皮肤衰老相关的信号通路,如参与刺激的检测、对类固醇激素的反应、水通道活性、肌肉收缩的调节、受体配体活性等。从PPI网络中共选出了10个核心基因,包括AQP4、TRPM8、TBR1、NTSR2、MPPED1、BARHL2、PAX9、CPN1、CES3和CHGB。值得注意的是,这些核心基因可能在皮肤衰老的过程和逆转中起关键作用。
作者为了探究核心基因在皮肤衰老中的重要作用,构建了“lncRNA-miRNA-mRNA”网络,识别出诸如“LOC100291323-hsa-mir-34a-5p-PAX9”和“LOC643339-hsa-mir-106b-5p-AQP4”等核心ceRNA机制。此外,利用CMAP平台筛选出10个显著的潜在抗皮肤衰老小分子,包括维生素A酸、吡非胺、司拉克丁、恩替诺特、溴他西尼、丁香酸、BRD-K9647565、依美斯汀、阿巴卡韦和鱼腾酮。维甲酸作为一种抗衰老皮肤药物已被研究者广泛讨论。吡非胺可以通过抑制p53的表达改善UVB致黑色素细胞衰老和色素沉着也曾被报道。总的来说,该研究揭示了皮肤衰老的潜在分子病理机制,并基于生物信息学的方法研究了逆转皮肤衰老的治疗药物。然而,其他小分子药物对皮肤衰老的作用和机制仍不清楚,需要更多的体内和体外实验进行验证。
原名:Identification of lncRNA-miRNA-mRNA Regulatory Network and Therapeutic Agents for Skin Aging by Bioinformatics Analysis.译名:通过生物信息学分析鉴定皮肤衰老相关的lncRNA-miRNA-mRNA调控网络和治疗药物。期刊:Biochemical GeneticsIF:2.4发表时间:2023年1月DOI号:10.1007/s10528-023-10334-8
18817128943

扫码关注 森煜智研微信号

扫码关注 森煜智研公众号